REMICADE®
JANSSEN-CILAG
Denominación genérica: Infliximab.
Forma farmacéutica y formulación: REMICADE Solución inyectable. Cada frasco ámpula contiene: Infliximab 100mg. Excipiente c.s. REMICADE contiene 100mg de Infliximab, un anticuerpo monoclonal quimérico IgG1, desarrollado de una línea celular recombinante cultivada por perfusión continua. REMICADE está disponible como polvo liofilizado estéril para infusión intravenosa. Después de reconstituirlo con 10 ml de agua estéril para inyección, cada ml contiene 10mg de Infliximab para diluir en solución de cloruro de sodio al 0.9%. No agitar.
Indicaciones terapéuticas: REMICADE está indicado en: Artritis reumatoide para: Disminución de signos y síntomas; así como mejorar la función física en: Pacientes con enfermedad activa, cuando la respuesta a fármacos modificadores de la enfermedad, incluyendo metotrexato, ha sido inadecuada. Pacientes con enfermedad moderada o gravemente activa, no tratados previamente con metotrexato. Prevención del daño articular estructural (erosiones y disminución del espacio articular). Mejorar función física de pacientes con enfermedad activa a pesar de tratamiento con metotrexato. Espondilitis Anquilosante para: Reducción de signos y síntomas. Mejoría de la función física. En pacientes con enfermedad activa. Artritis Psoriásica para: Disminución de signos y síntomas en pacientes con artritis psoriásica activa cuando la respuesta a fármacos modificadores de la enfermedad ha sido inadecuada. Mejorar la función física. Reducir la psoriasis evaluada por PASI (un índice que combina la evaluación del síntoma y el área de superficie corporal). En pacientes con artritis psoriásica activa. Psoriasis para: Disminución de signos y síntomas en pacientes con psoriasis y mejorar la calidad de vida en pacientes adultos, con placas psoriásicas moderadas y/o graves candidatos a tratamiento sistémico en quienes la fototerapia es inadecuada o inapropiada. Enfermedad de Crohn Pediátrica y en Adultos para: Reducción de signos y síntomas. Inducción y mantenimiento de la remisión clínica. Inducción de la cicatrización de la mucosa en adultos. Mejorar la calidad de vida. En pacientes con enfermedad de Crohn moderada a severa que han tenido respuesta inadecuada a las terapias convencionales. La terapia con REMICADE permite a los pacientes reducir o eliminar el uso de corticosteroides. Enfermedad de Crohn Fistulizante para: Reducción en el número de fístulas enterocutáneas y rectovaginales además el mantenimiento del cierre de ellas. Reducción de signos y síntomas y mejoría de la calidad de vida. Colitis Ulcerativa para: Reducir signos y síntomas. Inducir y mantener la remisión clínica. Inducir cicatrización de la mucosa. Mejorar la calidad de vida. Suspender o reducir la administración de corticoesteroides. Reducir las hospitalizaciones relacionadas con la colitis ulcerativa. En pacientes con colitis ulcerativa activa que han tenido una respuesta inadecuada a la terapia convencional. Eficacia clínica: Artritis reumatoide: La seguridad y eficacia de REMICADE fueron evaluadas en dos estudios pivote, multicéntricos, aleatorizados, doble ciego: ATTRACT (por sus siglas en inglés Anti-TNF Trial in Rheumatoid Arthritis with Concomitant Therapy) y ASPIRE (por sus siglas en inglés Active Controlled Study of Patients Receiving Infliximab for the Treatment of Rheumatoid Arthritis of Early Onset). Se permitio el uso concurrente de dosis estables de ácido fólico, corticoesteroides orales ( < 10mg/día) y/o antiinflamatorios no esteroideos. Los puntos primarios de desenlace fueron: reducción de signos y síntomas, evaluados por criterios de mejoría del Colegio Americano de Reumatología (ACR) (ACR20 para ATTRACT, y ACR-N a la semana 54 para ASPIRE), prevención del daño estructural y mejoría de función física. La reducción de signos y síntomas fue definida como una mejoría de al menos un 20% (ACR20) en la cuenta de articulaciones dolorosas e inflamadas, y en 3 de los siguientes 5 criterios: evaluación global por el médico; evaluación global por el paciente; mediciones de incapacidad/funcionalidad; escala visual análoga para dolor y velocidad de sedimentación globular o proteína C reactiva (PCR). ACR numérico utiliza los mismos criterios que ACR 20, se calcula tomando el menor porcentaje de mejoría en la cuenta de articulaciones inflamadas, cuenta de articulaciones dolorosas y la mediana del resto de los componentes de la respuesta de ACR. El daño estructural articular (erosiones y disminución del espacio articular) en manos y pies, fue evaluado por cambios radiológicos desde la evaluación basal con el índice de Sharp modificado (0-440). Los cambios en función física se midieron mediante el Cuestionario de Evaluación de Salud (HAQ; escala 0-3) al inicio y a las 102 semanas. El estudio ATTRACT evaluó respuesta a las 30 semanas (reducción de signos y síntomas), 54 semanas (prevención del daño estructural) y 102 semanas (mejoría de la función física) en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo de 428 pacientes con artritis reumatoide (AR) activa a pesar de tratamiento con metotrexato. Aproximadamente 50% de los pacientes estaban en clase funcional III. Los pacientes recibieron placebo, 3mg/Kg ó 10mg/Kg de REMICADE en las semanas 0, 2 y 6 y después cada 4 u 8 semanas. Todos los pacientes recibían dosis estables de metotrexato (promedio de 15mg/semana) 6 meses antes de iniciar el estudio y durante el mismo. En la semana 30, un porcentaje mayor de pacientes en todos los grupos que recibieron REMICADE, tuvo reducción significativa de signos y síntomas comparados con el grupo que recibió sólo metotrexato (Tabla 1). Esta respuesta fue observada en las primeras dos semanas, y fue mantenida durante 102 semanas de tratamiento (p < 0.001). En todos los pacientes que recibieron REMICADE se observó mejoría significativa (p < 0.05) en el número de articulaciones inflamadas y dolorosas, evaluación de dolor por el paciente, evaluación global de la enfermedad por el médico y el paciente, rigidez matutina, fatiga y PCR. Grados más elevados de respuesta clínica (ACR50 y ACR70) fueron observados con REMICADE tanto en las semanas 30, 54 y 102 de seguimiento. La prevención del daño estructural de la articulación (erosiones y disminución del espacio articular) fue observada en la semana 54 en todos los grupos de tratamiento con REMICADE (Tabla 1), y esta prevención fue detectada desde la semana 30 y mantenida hasta la semana 102 (p < 0.001). En la población de estudio, el 53% de los pacientes que recibieron REMICADE comparados con el 20% de los pacientes control se encontraron sin deterioro, es decir con un cambio ≤ 0 de la evaluación basal a la semana 54 según el índice de Sharp modificado. Se obtuvieron resultados similares para los componentes individuales del índice (erosiones y disminución del espacio articular). También se observó mejoría en función física (HAQ) en la semana 102 en pacientes tratados con REMICADE comparados con los pacientes control (Tabla 1) y fue observada desde la semana 54 (p < 0.001).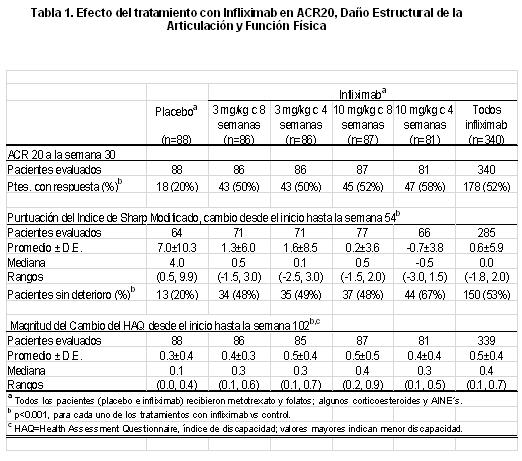
El estudio ASPIRE evaluó respuesta a las 54 semanas en 1,004 pacientes naïve a metotrexato con artritis reumatoide temprana (≤3 años de evolución). Los pacientes aleatorizados tenían una mediana de 51 años de edad, con una mediana del tiempo de evolución de 0.6 años, mediana de articulaciones inflamadas y dolorosas de 19 y 31 respectivamente. Todos los pacientes recibieron metotrexato (optimizado a 20mg semanales a la semana 8) y placebo ó 3mg/Kg ó 6mg/Kg de Infliximaben las semanas 0, 2 y 6 y después cada 8 semanas. Después de 54 semanas de tratamiento, ambas dosis de infliximab + metotrexato, obtuvieron mayor mejoría estadísticamente significativa de signos y síntomas al compararse con metotrexato solo, midiendo esta mejoría con la proporción de pacientes que alcanzaron ACR 20, 50 y 70. En los grupos que utilizaron infliximab + metotrexato, 15% alcanzaron una respuesta clínica importante vs. 8% en los pacientes tratados con metotrexato solo (p= 0.003). En el estudio ASPIRE, más del 90% de los pacientes tuvieron al menos dos radiografías evaluables. La inhibición de la progresión del daño estructural se observó en la semana 30 y 54 en los grupos tratados con infliximab + metotrexato comparados con el grupo de metotrexato solo. La combinación infliximab + metotrexato detuvo la progresión del daño articular en más pacientes al compararlos con el grupo de metotrexato solo, 97% vs. 86% respectivamente y mantuvo una mayor proporción, estadísticamente significativa de pacientes libres de erosiones 79% vs. 57%, respectivamente. Menos pacientes en los grupos de Infliximab + metotrexato (48%) desarrollaron erosiones en articulaciones no previamente afectadas comparados con el grupo de metotrexato solo (59%). Ambos grupos de tratamiento con Infliximab mostraron una mejoría estadísticamente significativa en el HAQ de la semana 54 con respecto al inicial, al compararlos con metotrexato solo: 0.7 en los grupos Infliximab + metotrexato vs. 0.6 para el grupo de metotrexato solo (p < 0.001). No se detectó empeoramiento de la evaluación mediante el índice resumido del componente mental del SF-36. Espondilitis Anquilosante: El estudio pivote fue un estudio multicéntrico, doble ciego, controlado con placebo que evaluó infliximab en 70 pacientes con espondilitis anquilosante activa severa. Durante la fase doble ciego de 3 meses, los pacientes recibieron 5mg/Kg de infliximab o placebo en las semanas 0, 2 y 6 (35 pacientes en cada grupo). A partir de la semana 12, los pacientes que recibieron placebo pasaron al grupo de infliximab y posteriormente todos los pacientes recibieron infliximab 5mg/Kg cada 6 semanas hasta la semana 54. Los resultados de este estudio fueron similares a los observados en 8 estudios iniciados por el investigador en 169 pacientes con espondilitis anquilosante activa. El tratamiento con infliximab produjo mejoría en los signos y síntomas, evaluada por el Índice de Actividad de la Espondilitis Anquilosante de Bath (BASDAI), ya que el 57% de los pacientes tratados con infliximab alcanzaron por lo menos un 50% de reducción de la puntuación basal de BASDAI, en comparación con el 9% de los pacientes tratados con placebo (p < 0.01). Se observó mejoría desde la semana 2, y fue mantenida hasta la semana 54. La función física fue evaluada utilizando el Bath Ankylosing Spondylitis Functional Index (BASFI) y el SF-36 comparado con placebo. Los pacientes tratados con infliximab mostraron mejoría significativamente mayor a la semana 12 en el BASFI y en la puntuación del componente físico del SF-36. Esta mejoría se mantuvo hasta la semana 54. Artritis Psoriásica: La eficacia y seguridad fueron evaluadas en un estudio multicéntrico doble ciego, controlado con placebo que evaluó infliximab en 104 pacientes con artritis psoriásica poliarticular activa. Durante las 16 semanas de la fase doble ciego, los pacientes recibieron 5mg/Kg o placebo, en las semanas 0, 2, 6 y 14 (52 pacientes en cada grupo). A la semana 16 se inició Infliximab con los pacientes del grupo placebo, subsecuentemente todos los pacientes recibieron 5mg/Kg de infliximab cada 8 semanas hasta la semana 46. El tratamiento con infliximab se asoció con mejoría de los signos y síntomas evaluados mediante criterios de ACR. El 65% de los pacientes alcanzaron ACR 20 a la semana 16, comparados con el 10% del grupo de pacientes tratados con placebo (p < 0.01). La respuesta fue similar no obstante el uso concomitante de metotrexato. La mejoría (ACR 20 y 50) fue observada tan rápido como a la semana 2 y se mantuvo hasta la semana 50 (ACR 20, 50 y 70). En la semana 16, el grupo tratado con infliximab tenía un promedio en DAS 28 (Disease Activity Score) de 3.0 (un DAS < 3.2 es considerado indicativo de baja actividad de la enfermedad), mientras que no se observó ningún cambio en el grupo con placebo (p < 0.01). Al final de la semana 50, ambas cohortes de sujetos tenían un puntaje similar de DAS28 indicando que el grupo tratado con Infliximab mantuvo el puntaje de DAS28 durante el tiempo, mientras que el grupo placebo presentó una disminución en DAS28 sólo después de haber cambiado a Infliximab. En el grupo de Infliximab se observó el 88.5% de respondedores evaluados con DAS28 a la semana 16 comparado con 25% en el grupo placebo (p < 0.01). Los Criterios de Respuesta en Artritis Psoriásica (PsARC) mostraron una rápida mejoría. A la semana 2, el 56% de sujetos en el grupo de Infliximab mostró mejoría versus 17% de pacientes en el grupo placebo (p < 0.01). A la semana 16 los resultados mostraron un 75% de respuesta en los pacientes tratados con Infliximab comparados con 21% en el grupo placebo (p < 0.01). En pacientes tratados con Infliximab, se observó disminución en los parámetros de actividad característicos de la artritis psoriásica (como el número de articulaciones inflamadas, dolorosas/hipersensibles, dactilitis y la presencia de entesopatía). En un subgrupo de pacientes con PASI (por sus siglas en inglés, Psoriasis Area and Severity Index) basal ≥2.5 se alcanzó una mejoría importante a la semana 16, con 68% (15/22) de los pacientes tratados con Infliximab que alcanzaron al menos 75% de mejoría respecto a su medición basal vs 0% (0/16) de los pacientes del grupo placebo, la mejoría se mantuvo hasta la semana 50. Los pacientes tratados con Infliximab mostraron mejoría en la capacidad funcional evaluada por HAQ (promedio de cambio de la evaluación a la semana 16 con respecto a la basal de 0.6 vs 0 del grupo tratado con placebo). Esta mejoría se mantuvo hasta la semana 50. Psoriasis: La eficacia del infliximab se evaluó en 2 estudios multicéntricos doble ciego, controlados con placebo: SPIRIT y EXPRESS. Los pacientes en ambos estudios tenían placas de psoriasis (área de superficie corporal (SC) ≥10% y PASI ≥ 12). El desenlace primario en ambos estudios fue el porcentaje de pacientes que alcanzaron ≥ 75% de mejoría en el PASI obtenido en la semana 10 comparado con el basal. Se clasificaron como respondedores notables a aquellos que obtuvieron ≥ 90% de mejoría en el PASI comparado con el basal. SPIRIT, evaluó la eficacia de infliximab como tratamiento de inducción a la remisión en 249 pacientes con placas de psoriasis que habían recibido previamente PUVA o tratamiento sistémico. Los pacientes recibieron infusiones de 3 ó 5mg/Kg de infliximab o placebo en las semanas 0, 2 y 6. Los pacientes con Valoración Global por el Médico (PGA, por sus siglas en inglés, Physician Global Asessment) con puntaje ≥ 3 fueron elegidos para recibir una aplicación adicional del mismo tratamiento a la semana 26. La proporción de pacientes con ≥75% de mejoría en PASI desde el basal (PASI 75) en la semana 10 fue 71.7% en el grupo de Infliximab 3mg/kg, 87.9% en el grupo de infliximab 5mg/kg y 5.9% en el grupo placebo (p < 0.001 para infliximab en comparación con el placebo). En la semana 10, una cantidad significativa de pacientes tratados con infliximab (3mg/kg: 45.5%; 5mg/kg: 57.6%), alcanzaron una respuesta marcada (≥90% de mejoría en PASI desde el basal) comparado con los pacientes tratados con placebo (2.0%). En el grupo de 3mg/kg, 60.6% de los pacientes mantuvieron la respuesta hasta la semana 14 y el 75.3% de los pacientes en el grupo de 5mg/kg mantuvieron la respuesta hasta la semana 18. En la semana 26 (veinte semanas después de la última dosis de inducción) 30% de los pacientes del grupo 5mg/kg y 13.8% de los pacientes en el grupo de 3mg/kg fueron respondieron en PASI 75, sugiriendo la necesidad de la terapia de mantenimiento. La calidad de vida relacionada con la salud, fue evaluada con el Índice Dermatológico de Calidad de Vida (DLQI, por sus siglas en inglés). El promedio basal de DLQI fue de 12, la mediana del cambio respecto a la evaluación inicial, en la semana 10, fue de -8.0 y -10.0 para los grupos de 3mg/Kg y 5mg/Kg de infliximab respectivamente, comparado con 0.0 en el grupo de pacientes tratados con placebo (p < 0.001 para ambos grupos vs placebo), demostrando así una mejoría sustancial en la calidad de vida de los pacientes tratados con Infliximab. EXPRESS, evaluó la eficacia de infliximab como tratamiento de inducción a la remisión y tratamiento de mantenimiento en 378 pacientes con placas de psoriasis que fueron candidatos a fototerapia o tratamiento sistémico. Los pacientes recibieron infusiones de 5mg/Kg de Infliximab o placebo en las semanas 0, 2 y 6, seguidas de infusiones cada 8 semanas a lo largo de 22 semanas en el grupo placebo y de 46 semanas en el grupo con infliximab. En la semana 24 el grupo placebo inició esquema de inducción con infliximab (5mg/Kg) seguido de tratamiento de mantenimiento (5mg/Kg). En EXPRESS la mediana de superficie corporal afectada basal fue de 29%, la mediana basal de PASI fue de 21.1 y la mayoría de los pacientes (89.9%) tenían un índice de PGA (Physician Global Assesment) de moderado, importante o severo. Antes de la terapia con Infliximab, 71.4% de los pacientes habían recibido tratamiento previo con PUVA, metotexato, ciclosporina o acitretina. A la semana 10, 80.4% de los pacientes tratados con Infliximab alcanzaron respuesta PASI 75 vs 2.6% en el grupo placebo (p < 0.001). La mediana de tiempo en alcanzar PASI 75 fue entre 2 y 6 semanas. La mejoría del PASI fue consistente en los diferentes subgrupos definidos por características demográficas, de la enfermedad y antecedentes de tratamiento al inicio del estudio. Se alcanzó respuesta importante (PASI 90) en el 57.1 % del grupo con Infliximab comparado con 1.3% en el grupo placebo (p < 0.001), a la semana 10. La respuesta se mantuvo durante las 24 semanas del periodo controlado con placebo. Las tasas de respuesta en el PASI durante 50 semanas se presentan en la tabla 2.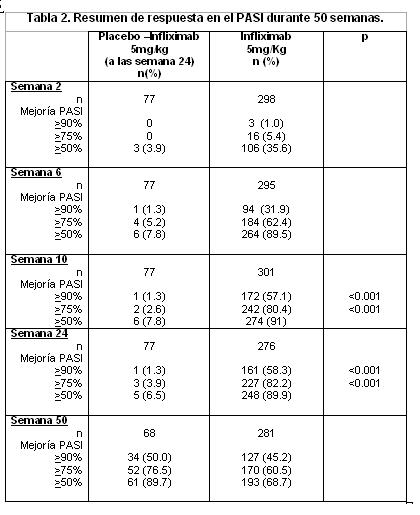
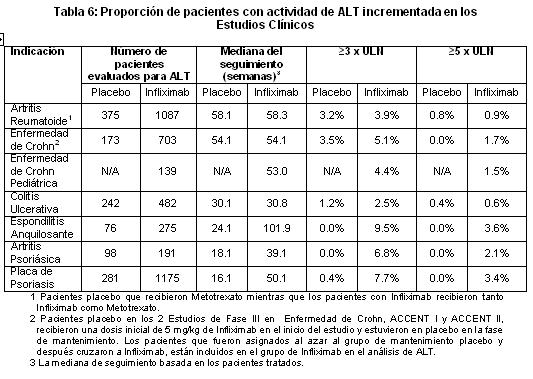
A la semana 10, 82.9% de los pacientes alcanzaron un PGA mínimo o nulo comparado con 3.9% del grupo placebo (p < 0.001). La calificación de PGA a las semanas 6, 10,24 y 50 se presentan en la tabla 3.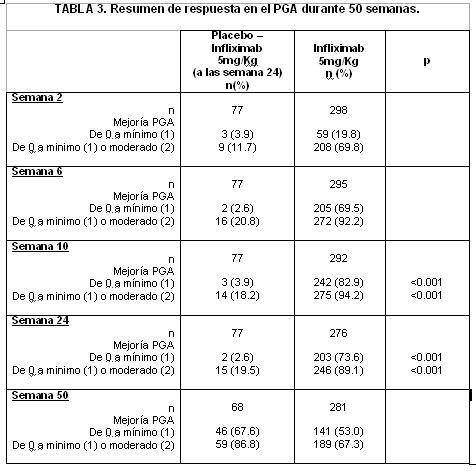
La mediana basal en el DLQI fue de 12.5. El promedio basal en el SF36 fue de 45.6 en el componente físico SF-36 y 45.7 en el componente mental. La calidad de vida mejoró significativamente al comparar con placebo a las semanas 10 y 24 cuando se evaluaron ambos índices (DLQI y SF-36). La mediana basal del índice ungueal para psoriasis (NAPSI) fue de 4 y el promedio de uñas afectadas por psoriasis fue de 10. Los pacientes tratados con infliximab mostraron una clara mejoría de la psoriasis de la uña basal comparados con los pacientes del grupo placebo, medida por NAPSI y por disminución en el numero de uñas afectadas. Enfermedad de Crohn en pacientes adultos: La seguridad y eficacia de dosis única y múltiples de REMICADE se evaluaron en dos estudios aleatorizados, doble ciego, controlados con placebo, en pacientes con enfermedad de Crohn activa moderada a severa (Índice de actividad de enfermedad de Crohn (CDAI) ≥220, ≤400) y con respuesta inadecuada a terapias convencionales. Se permitió uso concurrente de tratamientos convencionales a dosis estables y 92% de pacientes continuaron recibiendo dichos tratamientos. En el estudio de una sola dosis en 108 pacientes, 22/27 (81%) pacientes tratados con 5mg/Kg de Infliximab lograron respuesta clínica (Disminución en el CDAI ≥ 70 puntos), comparados con 4/25 (16%) pacientes con placebo (p < 0.001). También en la semana 4, 13/27 (48%) pacientes que recibieron tratamiento con Infliximab lograron una remisión clínica (CDAI < 150), comparados con 1/25 (4%) que recibieron placebo. En el estudio multidosis, 573 pacientes recibieron 5mg/Kg en la semana 0 y luego fueron aleatorizados a tres grupos de tratamiento de mantenimiento. Un grupo recibió placebo en las semanas 2 y 6 y posteriormente cada 8 semanas, el segundo grupo recibió 5mg/Kg de Infliximab en las semanas 2 y 6 y luego cada 8 semanas y un tercer grupo recibió 5mg/Kg de Infliximab en las semanas 2 y 6 y posteriormente 10mg/Kg de Infliximab cada 8 semanas. Los pacientes considerados respondedores en la semana 2 fueron aleatorizados y analizados de manera separada de los no respondedores. A la semana 2, 58% (335/573) de los pacientes presentaban respuesta clínica (disminución en el CDAI > 25% y > 70 puntos). Una proporción significativamente mayor de los pacientes en los grupos de mantenimiento con 5mg/Kg y 10mg/Kg alcanzó remisión clínica en la semana 30, en comparación con los pacientes del grupo de mantenimiento con placebo (p < 0.001). La mediana del tiempo hasta la pérdida de respuesta fue de 46 semanas para el grupo de tratamiento de mantenimiento con Infliximab, contra 19 semanas para el grupo de tratamiento de mantenimiento con placebo. A los pacientes que alcanzaron respuesta y que posteriormente la perdieron, se les permitió recibir el tratamiento episódico con Infliximab a una dosis de 5mg/Kg más que la dosis de mantenimiento que se les había asignado de manera aleatoria. El 89% (50/56) de los pacientes que perdieron la respuesta clínica con una dosis de mantenimiento de 5mg/Kg de Infliximab cada 8 semanas, respondieron a la infusión de 10mg/Kg de Infliximab. Se observó mejoría significativa en la calidad de vida evaluada por SF-36 y el Cuestionario de Enfermedad Inflamatoria Intestinal específico para la enfermedad (IBDQ) (p < 0.001) en los pacientes tratados con Infliximab a la semana 30. Para los pacientes que recibían corticoesteroides en la visita basal, la proporción de estos pacientes en remisión clínica y sin recibir corticoesteroides en la semana 30 fue del 31% para el grupo de mantenimiento con 5mg/Kg y del 37% para el grupo de mantenimiento con 10mg/Kg, en comparación con el 11% de pacientes en el grupo de mantenimiento con placebo (p=0.001 para los grupos de Infliximab 5mg/Kg y 10mg/Kg). La mediana de la dosis de corticoesteroides en la visita basal (20mg/día) se redujo a 10mg/día en el grupo de mantenimiento con placebo y a 0mg/día en los grupos de mantenimiento con Infliximab en la semana 30, lo que demuestra que al menos el 50% de los pacientes en mantenimiento con Infliximab fueron capaces de suspender el uso de esteroides. A la semana 10 una proporción significativamente mayor de pacientes de los grupos combinados de mantenimiento con Infliximab (31%) presentó cicatrización de la mucosa en comparación con los pacientes del grupo de placebo (0%, p=0.010). Los resultados fueron similares a la semana 54. Enfermedad de Crohn fistulizante: Se evaluaron seguridad y eficacia de Infliximab en un estudio aleatorizado, doble-ciego, controlado con placebo de 94 pacientes con enfermedad de Crohn fistulizante, que tuvieran fístulas de por lo menos 3 meses de evolución. En total, 31 pacientes recibieron tratamiento con REMICADE (5mg/Kg). Aproximadamente 93% de los pacientes habían recibido previamente tratamiento con antibióticos o inmunosupresores. Se permitió el uso concurrente de terapias convencionales a dosis estable y 83% de los pacientes continuaron recibiendo al menos uno de estos medicamentos. Los pacientes recibieron tres dosis de placebo o REMICADE en la semana 0, 2 y 6. Se les dio seguimiento por 26 semanas. El desenlace primario fue la proporción de pacientes que tuvieron respuesta clínica, definida como ≥50% de reducción del número de fístulas que drenaban al comprimirse levemente, en por lo menos dos visitas consecutivas (4 semanas), sin incremento en medicamentos o necesidad de cirugía para enfermedad de Crohn. El 68% (21/31) de los pacientes tratados con 5mg/Kg de REMICADE tuvieron respuesta clínica versus 26% (8/31) de pacientes tratados con placebo (p=0.002). El inicio de respuesta en el grupo tratado con REMICADE fue de una mediana de 2 semanas. La duración de la respuesta fue en promedio de 12 semanas. Adicionalmente el cierre de las fístulas fue observado en 55% de pacientes tratados con REMICADE comparado con 13% de tratados con placebo (p=0.001). La seguridad y eficacia de infusiones repetidas de Infliximab en pacientes con enfermedad de Crohn fistulizante fue estudiada en un ensayo clínico de un año. Un total de 306 pacientes recibieron 3 dosis de Infliximab de 5mg/Kg en las semanas 0,2 y 6. De los pacientes aleatorizados al inicio, 87% tenían fístula perianal, 14% fístula abdominal y 9% fístula rectovaginal. La mediana del puntaje de CDAI (Crohn's Disease Activity Index) fue de 180. Ciento noventa y cinco pacientes respondieron a la 3ra dosis (ver definición primaria de respuesta de enfermedad fistulizante arriba), a la semana 14 se aleatorizaron para recibir placebo ó 5mg/Kg de Infliximab cada 8 semanas hasta la semana 46. En la semana 14, 65% (177/273) de los pacientes asignados alcanzaron respuesta al tratamiento. Los pacientes asignados al esquema de mantenimiento con Infliximab mantuvieron significativamente mayor tiempo la respuesta comparado con los asignados a placebo (p=0.001). La mediana de tiempo de mantenimiento de la respuesta fue > 40 semanas en el grupo de Infliximab comparado con 14 semanas en el grupo placebo. A la semana 54, el 38% de los pacientes tratados con REMICADE no tenían fístulas drenando comparado con el 22% (20/90) del grupo tratado con placebo (p=0.02). El grupo tratado con Infliximab mostró mayor mejoría respecto al basal en el puntaje de CDAI comparado con el del grupo placebo, (p=0.04). Los pacientes que alcanzaron respuesta de la fístula y la perdieron subsecuentemente fueron elegibles para recibir REMICADE, 66% (25/38) respondieron a 5mg/Kg de REMICADE, y 57% (12/21) de los pacientes respondieron a dosis de 10mg/Kg de REMICADE como terapia de mantenimiento. Comparados con el grupo placebo, los pacientes del grupo tratado con REMICADE tuvieron una tendencia a tener menos hospitalizaciones. Enfermedad de Crohn en pacientes pediátricos: La seguridad y eficacia de dosis únicas y múltiples de REMICADE fueron evaluadas en un estudio fase II, multicéntrico de dosis única que incluyó 21 pacientes pediátricos con enfermedad de Crohn activa y en un estudio fase III multicentrico de múltiple aleatorizado en 112 pacientes pediátricos con enfermedad de Crohn activa (estudio REACH). En el estudio Fase II de dosis única en 21 pacientes (11 a 17 años de edad, mediana de la edad 15.0 años), todos los pacientes alcanzaron una respuesta clínica (disminución en CDAI ≥70 puntos o una disminución de PCDAI ≥10) en algunos puntos en las 20 semanas siguientes a la dosis única de Infliximab, y una remisión clínica (definida como una reducción en la puntuación de CDAI modificada por debajo de 150 puntos o una reducción en PCDAI debajo de 10) en 10 pacientes (47.6%). De las 3 dosis administradas (1, 5 o 10mg/kg), en los grupos de tratamiento de 5mg/kg y de 10mg/kg hubo más pacientes que alcanzaron la remisión clínica (16.7% en el grupo de tratamiento con dosis de Infliximab de 1mg/kg comparado con 57.1% y 62.5% en los grupos de tratamiento con dosis de Infliximab de 5mg/kg y 10mg/kg, respectivamente). En los 7 pacientes que tuvieron enfermedad fistulizante, las fístulas se cerraron por lo menos en 1 visita de evaluación (8 semanas). En un estudio Fase III de dosis múltiple (REACH), 112 pacientes (6 a 17 años, mediana de edad 13.0 años) recibieron 5mg/kg de Infliximab en las semanas 0, 2 y 6. Los pacientes evaluados por el investigador para estar en respuesta clínica en la Semana 10, se asignaron aleatoriamente y recibieron Infliximab 5mg/kg cada 8 semanas o cada 12 semanas como un régimen de mantenimiento. Se permitió el cruce de una dosis más alta o un intervalo de dosificación más corto en caso de que la respuesta se perdiera durante el tratamiento de mantenimiento. En REACH, la respuesta clínica en la semana 10 fue 88.4% (99/112) comparado con 66.7% (128/192) en adultos (ACCENT 1). De manera similar, la proporción de pacientes que alcanzaron remisión clínica en la Semana 10 fue 58.9% (66/112) comparado con 39.1% (75/192) en adultos (ACCENT 1). En la Semana 30, la proporción de pacientes con respuesta clínica fue significativamente más alto en el grupo de tratamiento de cada 8 semanas (73.1%, 38/52) que en el grupo de tratamiento de mantenimiento de cada 12 semanas (47.1%, 24/51; p= 0.007). En la semana 54, la proporción de pacientes con respuesta clínica, también fueron significativamente más para el grupo de tratamiento de de cada 8 semanas (63.5%, 33/52) que en el grupo de tratamiento de cada 12 semanas (33.3%, 17/51; p = 0.002). En la semana 30, la proporción de pacientes en remisión clínica fue significativamente más alto en el grupo de tratamiento de cada 8 semanas (59.6%, 31/52) que en el grupo de tratamiento de cada 12 semanas (35.3%, 18/51; p = 0.013). En la semana 54, la proporción de pacientes en remisión clínica también fue significativamente más alto para pacientes en los grupos de tratamiento de cada 8 semanas (55.8%, 29/52) que en el de cada 12 semanas (23.5%, 12/51; p < 0.001). En REACH, el cambio desde el inicio en el uso promedio diario de Corticoesteroides fue significativo en las Semanas 10, 30 y 54 (p < 0.001). Para los pacientes que recibian Corticoesteroides en el inicio, la remisión clínica en REACH alcanzada sin Corticoesteroides en la Semana 30 fue de 45.8% para el grupo de tratamiento de cada 8 semanas y 33.3% para el grupo de tratamiento de cada 12 semanas. En la Semana 54, 45.8% de los pacientes en el grupo de tratamiento de cada 8 semanas y 16.7% de pacientes en el grupo de tratamiento de cada 12 semanas tuvieron remisión clínica y no recibieron Corticoesteroides. La calidad de vida se evaluó usando la puntuación IMPACT III (un cuestionario QOL desarrollado y validado específicamente para pacientes pediátricos con enfermedad inflamatoria del intestino). Éste se aplicó solamente a los pacientes de Norte América. Los cambios promedio (un cambio negativo indica mejoría) fueron significativos (p < 0.001) desde la basal de la puntuación IMPACT III en las Semanas 10, 30 y 54 (-22.9, -21.1 y -24.3, respectivamente). El índice de altura z, es una medida de la desviación de la altura esperada en una población de la misma edad y género en los pacientes pediátricos. En la población estudiada, la mediana del índice de altura z en la basal fue de -1.6. El cambio promedio desde la basal en el índice de altura z, fue de 0.3 y 0.4 para la semana 30 y la semana 54, respectivamente. El índice de altura z fue mejorado significativamente desde la basal en ambas semanas, semana 30 (p < 0.001) y semana 54 (p < 0.001). Colitis Ulcerativa: La seguridad y la eficacia de Infliximab fueron evaluadas en dos estudios (ACT 1 y ACT 2) aleatorios, doble ciego, controlados con placebo en pacientes adultos con colitis ulcerativa activa de moderada a severa (Índice de Mayo 6 a 12; subíndice de endoscopia ≥2) con una respuesta inadecuada a terapias convencionales [corticoesteroides orales, aminosalicilatos y/o inmunomoduladores (6-MP, AZA)]. Se permitieron dosis estables concomitantes de corticoesteroides orales, aminosalicilatos, y/o agentes inmunomoduladores. En ambos estudios, los pacientes fueron aleatorizados para recibir placebo, 5mg/Kg de Infliximab, o 10mg/Kg de Infliximab en las semanas 0, 2, 6, 14 y 22. La disminución de corticoesteroides se permitió después de la semana 8. En ambos estudios, un porcentaje significativamente mayor de pacientes en los grupos con Infliximab tuvo respuesta y remisión clínica en la semana 8 al compararse con el placebo. Además, tanto en ACT1 como en ACT 2, una proporción considerablemente mayor de pacientes tratados con 5mg/Kg o 10mg/Kg de Infliximab, experimentaron respuesta y remisión clínica en la semana 30 al compararse con placebo. Además, la proporción de pacientes con respuesta sostenida (es decir, respuesta clínica tanto en la semana 8 como en la 30) en los grupos de Infliximab fue al menos dos veces mayor comparado con el grupo de placebo. El resultado de las semanas 8 y 30 se muestran en la tabla 4. De los pacientes tratados con corticoesteroides al inicio, una proporción significativamente mayor de los pacientes con Infliximab alcanzó remisión clínica en la semana 30 y fueron capaces de suspender los corticoesteroides, al compararlos con los pacientes tratados con placebo (22.3% vs 7.2% respectivamente, ver tabla 4). Adicionalmente, en las semanas 8 y 30, una proporción significativamente mayor de pacientes con dosis de 5mg/Kg y 10mg/Kg en el ACT 1 y ACT 2 alcanzó la cicatrización de la mucosa al compararse con pacientes del grupo placebo. La proporción de pacientes con cicatrización de la mucosa fue similar en los 2 grupos de dosis de Infliximab en los dos estudios (ver tabla 4). Infliximab mejoró la Calidad de Vida, confirmado por mejoría significativa en ambas medidas específicas de la enfermedad, IBDQ (Inflammatory Bowel Disease Questionnaire) y SF-36. Desde el inicio hasta la semana 30 en los datos reunidos del ACT 1 y el ACT 2, el número promedio de hospitalizaciones fue 50 % más bajo en el grupo de tratamiento con Infliximab que en el grupo tratado con placebo (9 contra 18 hospitalizaciones por cada 100 pacientes, p = 0.005). No se observaron diferencias notables entre los grupos de tratamiento con 5mg/Kg y 10mg/Kg de Infliximab.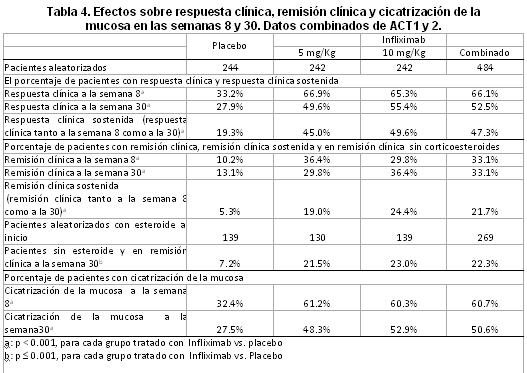
Farmacocinética y farmacodinamia: REMICADE (Infliximab) es un anticuerpo monoclonal quimérico humano-murino que se une con alta afinidad a las formas soluble y transmembranal del TNF-a, pero no a la linfotoxina (TNFb). En bioensayos in vitro Infliximab inhibe la actividad funcional del TNF-. Infliximab previene enfermedad en ratones transgénicos que desarrollan poliartritis como resultado de una expresión constitutiva del TNF-a y cuando se administra una vez desarrollada, permite que las articulaciones erosionadas se reparen. In vivo Infliximab forma rápidamente complejos estables con el TNF-a, proceso relacionado con una disminución en la bioactividad del TNF-a. La evaluación histológica de biopsias de colon obtenidas antes y 4 semanas después de la administración de REMICADE revelaron una disminución del TNF-a detectable. El tratamiento con REMICADE en la enfermedad de Crohn, se asoció con disminución de marcadores de inflamación como la PCR, comúnmente elevado. El número de leucocitos en sangre periférica se afectó mínimamente en pacientes con REMICADE, aunque se detectaron desviaciones a la izquierda en linfocitos, monocitos y neutrófilos. Se ha observado una disminución en respuestas proliferativas por células mononucleares de sangre periférica (CMSP) de pacientes tratados con Infliximab, sin embargo no se han observado cambios sustanciales en producción de citocinas por parte de las CMSP estimulados. El análisis de células mononucleares de lámina propia obtenidas por biopsia de mucosa intestinal, demuestra que el tratamiento con Infliximab causa una reducción en el número de células capaces de expresar TNF-a e interferón-c Estudios histológicos adicionales, proveen evidencia de que el tratamiento con REMICADE disminuye la infiltración de células inflamatorias a áreas afectadas del intestino y la presencia de marcadores de inflamación en estos sitios. Se ha detectado TNF-a en concentraciones elevadas en articulaciones de pacientes con Artritis Reumatoide (AR) y correlacionan con actividad de la enfermedad. También se han encontrado concentraciones elevadas de TNF-a en el líquido articular, tejidos y lesiones cutáneas en pacientes con artritis psoriásica. En Artritis Reumatoide (AR) el tratamiento con REMICADE reduce la infiltración de células inflamatorias dentro de áreas afectadas de las articulaciones, así como expresión de moléculas mediadoras de adhesión celular, quimioatrayentes y degradación de tejidos. Después del tratamiento con REMICADE, los pacientes presentaron una disminución en los niveles séricos de Interleucina 6 (IL-6) y proteína C reactiva (PCR) comparados con determinaciones iniciales. Los linfocitos periféricos no presentaron disminución significativa en número ni en respuesta proliferativa a la estimulación mitogénica in vitro comparados con células de pacientes no tratados. En los pacientes con psoriasis, el tratamiento con Infliximab resultó en una disminución de la inflamación epidérmica y en la normalización de la diferenciación de queratinocitos en las placas psoriásicas. Farmacocinética: Los datos de un estudio con infusión intravenosa única de 1, 3, 5, 10 ó 20mg/Kg de REMICADE mostraron una relación lineal y directa con: dosis administrada, concentración sérica máxima (Cmax) y área bajo la curva (AUC). El volumen de distribución en estado estable (mediana de Vd de 3 a 4.1 litros) no fue dependiente de la dosis administrada e indicó que REMICADE es predominantemente distribuido en el compartimiento vascular. Se observó una farmacocinética no dependiente del tiempo. La vía de eliminación de REMICADE no ha sido caracterizada. No se encontraron diferencias en la depuración o en el volumen de distribución en subgrupos de pacientes definidos por edad o peso, función renal o hepática. No se observaron diferencias notables en los parámetros farmacocinéticos tras dosis única entre pacientes pediátricos y adultos con enfermedad de Crohn. Con dosis únicas de 3, 5, y 10mg/Kg; los valores farmacocinéticos medios para Cmax resultaron en 77, 118 y 277 g/ml respectivamente. El promedio de vida media terminal para estas osciló entre 8 a 9.5 días. En la mayoría de pacientes, el Infliximab pudo ser detectado en suero durante al menos 8 semanas después de una infusión única. Después del régimen recomendado de 3 dosis, se observó una ligera acumulación de Infliximab en suero después de la segunda dosis y ninguna acumulación posterior clínicamente relevante a partir de entonces. En la mayoría de pacientes con enfermedad de Crohn fistulizante, el Infliximab fue detectado en suero por 12 semanas (rango de 4 a 28 semanas) después de la administración del régimen.
Contraindicaciones: REMICADE no debe administrarse a pacientes con hipersensibilidad conocida a Infliximab, proteínas murinas o a cualquiera de sus excipientes. REMICADE está contraindicado en pacientes con infecciones severas, como tuberculosis, sepsis, abscesos o infecciones oportunistas. REMICADE está contraindicado en pacientes con insuficiencia cardiaca de moderada a severa (Clase III/IV de la New York Heart Association -NYHA- por sus siglas en inglés). (Ver Precauciones y reacciones secundarias y adversas).
Precauciones generales: Reacciones de hipersensibilidad y reacciones a la infusión: REMICADE se asocia con reacciones agudas e hipersensibilidad tardía a la infusión. Éstas, difieren en tiempo de inicio, por lo tanto, todo paciente que reciba REMICADE debe estar bajo observación por lo menos 1 hora, después de la infusión. Para minimizar la incidencia de las reacciones de hipersensibilidad, incluyendo las reacciones a la infusión y reacciones parecidas a la enfermedad del suero, REMICADE deberá ser administrado como una terapia regular de mantenimiento después del régimen de inducción a las semanas 0, 2 y 6 (ver Dosis y Vía de administración). Reacciones agudas durante la infusión, pueden presentarse inmediatamente o dentro de las primeras horas después de la infusión. Si ocurre una reacción aguda a la infusión, la administración debe ser interrumpida inmediatamente. Algunos de estos efectos se han descrito como anafilaxia. Deberán estar disponibles para