REPATHA
AMGEN
Denominación genérica: Evolocumab.
Forma farmacéutica y formulación: La solución es entre transparente y opalescente, entre incolora y amarillenta, y está prácticamente libre de partículas. Cada jeringa prellenada (PFS) o pluma precargada contiene 140 mg de evolocumab en 1 mL de solución. Evolocumab 140 mg Excipientes c.s. REPATHA es un anticuerpo monoclonal IgG2 humano de origen ADN recombinante expresado en células de ovario de hámster chino (CHO).
Indicaciones terapéuticas: Prevención de eventos cardiovasculares: REPATHA está indicado en combinación con otras terapias en adultos con enfermedad cardiovascular establecida, para reducir el riesgo de infarto de miocardio, enfermedad vascular cerebral isquémica y revascularización coronaria. Hipercolesterolemia y Dislipidemia Mixta: REPATHA está indicado en adultos con hipercolesterolemia primaria (heterocigoto familiar y no familiar) o dislipidemia mixta como complemento a la dieta.En combinación con una estatina o estatina con otras terapias de reducción de lípidos en pacientes que no consiguen alcanzar los niveles objetivo de colesterol de baja densidad (LDL-C por sus siglas en inglés) con la dosis máxima tolerada de estatina, o bien, Solo o en combinación con otras terapias de reducción de lípidos en pacientes que son intolerantes a las estatinas, o en los que las estatinas están contraindicadas. Hipercolesterolemia Familiar Homocigota: REPATHA está indicado en adultos y adolescentes desde los 12 años de edad con hipercolesterolemia familiar homocigota (HoFH, por sus siglas en inglés) en combinación con otras terapias de reducción de lípidos.
Farmacocinética y farmacodinamia: Grupo fármaco-terapéutico: Otros agentes modificadores de los lípidos. Código ATC: C10AX13. Farmacocinética:Absorción y Distribución: Después de una dosis única subcutánea de 140 mg o 420 mg de REPATHA administrada a adultos sanos, la mediana de concentraciones séricas pico fue alcanzada en 3 a 4 días. La administración de una única dosis subcutánea de 140 mg provocó una Cmáx media (DE) de 13.0 (10.4) mg/mL y una ABCfinal media (DE) de 96.5 (78.7) día•mg/mL. La administración de una dosis subcutánea única de 420 mg provocó una Cmáx media (DE) de 46.0 (17.2) mg/mL y una ABCfinal media (DE) de 842 (333) día•mg/mL. Tres dosis subcutáneas de 140 mg fueron bioequivalentes a una dosis subcutánea única de 420 mg. A partir de modelos farmacocinéticos, se determinó que la biodisponibilidad absoluta tras la administración SC fue del 72%. Después de una dosis intravenosa única de 420 mg de REPATHA, el volumen de distribución promedio (DE) en estado de equilibrio fue estimado en 3.3 (0.5) L, lo que sugiere que evolocumab tiene una limitada distribución tisular. Metabolismo y Excreción: Biotransformación: REPATHA está compuesto únicamente de aminoácidos y carbohidratos como las inmunoglobulinas naturales y es improbable que se elimine a través de mecanismos metabólicos hepáticos. Se prevé que su metabolismo y eliminación sigan las vías de aclaramiento de las inmunoglobulinas, que se degradan en pequeños péptidos y aminoácidos simples. Eliminación: Se calculó que evolocumab tiene una vida media efectiva de entre 11 a 17 días. En pacientes con hipercolesterolemia familiar heterocigota (HFHe) que recibían dosis altas de estatinas, la exposición sistémica a evolocumab fue ligeramente inferior que en sujetos que recibían dosis de estatinas entre bajas y moderadas (la relación de ABCfinal 0.74 [IC 90% 0.29; 1.9]). El aumento de aproximadamente el 20% en el aclaramiento se debe en parte a que las estatinas aumentaron la concentración de PCSK9, lo que no afectó negativamente al efecto farmacodinámico de evolocumab en los lípidos. Los análisis farmacocinéticos de la población determinaron que no existían diferencias apreciables en las concentraciones séricas de evolocumab en los pacientes con hipercolesterolemia (hipercolesterolemia no familiar o hipercolesterolemia familiar) que tomaban estatinas de forma concomitante. Linealidad/No Linealidad: Después de una dosis intravenosa única de 420 mg, la media (DE) de depuración sistémica se estimó en 12 (2) mL/h. En estudios clínicos con dosificación subcutánea repetida durante 12 semanas, se observaron incrementos en la exposición proporcionales a la dosis con regímenes de dosificación de 140 mg y mayores. Se observó una acumulación aproximada de dos a tres veces en concentraciones séricas mínimas (Cmín [DE] 7.21 [6.6]) después de dosis de 140 mg cada 2 semanas o después de dosis de 420 mg administradas mensualmente (Cmín [DE] 11.2 [10.8]), y concentraciones séricas mínimas cercanas al estado e equilibrio a las 12 semanas de dosificación. No se observaron cambios dependientes del tiempo en las concentraciones séricas durante un periodo de 124 semanas. Poblaciones Especiales: Insuficiencia Renal: Como no se sabe que los anticuerpos monoclonales se eliminen a través de vías renales, no se espera que la función renal afecte la farmacocinética de evolocumab. En un ensayo clínico de 18 pacientes con función renal normal (tasa estimada de filtración glomerular [TFGe] ≥ 90 mL/min/1.73 m2, n = 6), insuficiencia renal grave (TFGe < 30 mL/min/1.73 m2, n = 6), o enfermedad renal en etapa terminal (ESRD) que recibían hemodiálisis (n = 6), la exposición a evolocumab después de una sola dosis subcutánea de 140 mg disminuyó en pacientes con insuficiencia renal grave o ESRD que recibían hemodiálisis. Las reducciones en los niveles de PCSK9 en pacientes con insuficiencia renal grave o ESRD que recibían hemodiálisis fueron similares a aquellos con función renal normal. (Ver uso en Poblaciones Específicas)(8.6).Insuficiencia Hepática: No se requiere ajuste de la dosis en pacientes con insuficiencia hepática leve (Child-Pugh clase A). La dosis única subcutánea de 140 mg de REPATHA fue estudiada en 8 pacientes con insuficiencia hepática leve, 8 pacientes con insuficiencia hepática moderada, y 8 sujetos sanos. Se encontró que la exposición a evolocumab es aproximadamente 40% a 50% inferior comparada con sujetos sanos. Sin embargo, se encontró que los niveles basales de PCSK9 y el grado y duración de la neutralización de la PCSK9 son similares entre pacientes con insuficiencia hepática leve a moderada y voluntarios sanos. Esto resultó en valores similares tanto para la reducción absoluta de LDL-C como para el intervalo de tiempo necesario para dicha reducción. REPATHA no se ha estudiado en pacientes con insuficiencia hepática severa (Child-Pugh clase C) (ver Precauciones generales). Peso Corporal: El peso corporal fue una covariable significativa en el análisis farmacocinético de la población afectando a las concentraciones mínimas de evolocumab, sin embargo no hubo impacto en la reducción del LDL-C. Tras la administración subcutánea repetida de 140 mg cada 2 semanas, las concentraciones mínimas en la semana 12 fueron de un 147% mayores y un 70% menores en pacientes de 69 kg y 93 kg, respectivamente, que la de un paciente estándar de 81 kg. Se observó una menor influencia del peso corporal con dosis mensuales subcutáneas repetidas de 420 mg de evolocumab. Otras Poblaciones Especiales: Los análisis farmacocinéticos de la población sugieren que no es necesario ajustar la dosis en función de la edad, la raza o el sexo. La farmacocinética de evolocumab fue influenciada por el peso corporal, pero este hallazgo no tuvo ningún efecto notable en la reducción del LDL-C. Por lo tanto, no es necesario ajustar la dosis en función del peso corporal. Efectos sobre la Capacidad para Conducir y Usar Maquinaria: La influencia de REPATHA sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Farmacodinamia: Mecanismo de Acción: Evolocumab se enlaza selectivamente a PCSK9 y previene la unión de la PCSK9 circulante con el receptor de lipoproteína de baja densidad (LDLR) localizado sobre la superficie celular del hígado, y de esta forma previene la degradación del LDLR mediada por PCSK9. El incremento en los niveles de LDLR del hígado resulta en reducciones asociadas de LDL-C sérico. Efectos Farmacodinámicos: En estudios clínicos, REPATHA redujo la PCSK9 libre, el LDL-C, el colesterol total (TC), la apolipoproteína B (ApoB), el colesterol no HDL (C-no-HDL), los índices TC/HDL-C y ApoB/ApoA1, el colesterol de muy baja densidad (VLDL-C), los triglicéridos (TG), y la Lp(a) e incrementó el colesterol de alta densidad (HDL-C) y la apolipoproteína A1 (ApoA1) en pacientes con hipercolesterolemia primaria, dislipidemia mixta y enfermedad cardiovascular establecida. Una administración subcutánea única de REPATHA de 140 mg o 420 mg resultó en supresión máxima de la PCSK9 circulante no-ligada por 4 horas seguida de una reducción en el LDL-C alcanzando una media del nadir de la respuesta de 14 y 21 días, respectivamente. Los cambios en la PCSK9 no-ligada y las lipoproteínas séricas fueron reversibles a la discontinuación de REPATHA. No se observó incremento en la PCSK9 no-ligada o el LDL-C arriba del valor basal durante el periodo de lavado de evolocumab lo que sugiere que los mecanismos compensatorios para incrementar la producción de PCSK9 y LDL-C no ocurren durante el tratamiento. Los regímenes subcutáneos de 140 mg cada 2 semanas y 420 mg una vez por mes fueron equivalentes en la reducción promedio de LDL-C (media de 10 y 12 semanas), con un resultado de -72% a -57% con respecto a los valores basales en comparación con placebo. El tratamiento con REPATHA resultó en reducciones similares de LDL-C cuando se usó solo o en combinación con otras terapias de reducción de lípidos. El efecto de reducción del LDL-C es sostenido; la duración más prolongada medida fue de 112 semanas. Eficacia Clínica: Prevención de eventos cardiovasculares: El Estudio FOURIER fue un estudio doble ciego, aleatorizado, controlado con placebo, controlado por evento en 27,564 (13,784 REPATHA, 13,780 placebo) pacientes adultos con enfermedad cardiovascular establecida y con LDL-C ≥ 70 mg/dL y/o HDL-C ≥ 100 mg/dL a pesar del tratamiento con estatinas de intensidad alta o moderada. Los pacientes fueron asignados aleatoriamente 1: 1 para recibir inyecciones subcutáneas de REPATHA (140 mg cada 2 semanas o 420 mg una vez al mes) o placebo; el 86% usó el régimen de cada 2 semanas a lo largo del estudio. La mediana de seguimiento fue de 26 meses. En general, el 99.2% de los pacientes fueron seguidos hasta el final del estudio o la muerte. La edad media (DE) al inicio del estudio fue de 63 (9) años, con 45% con al menos 65 años de edad; 25% eran mujeres. La población de prueba fue 85% blanca, 2% negra y 10% asiática; 8% identificado como etnia hispana. En cuanto a los diagnósticos previos de enfermedades cardiovasculares, el 81% tenía infarto de miocardio previo, el 19% tenía ictus no hemorrágico previo y el 13% tenía enfermedad arterial periférica sintomática. Los factores de riesgo iniciales adicionales seleccionados incluyeron hipertensión (80%), diabetes mellitus (1% tipo 1, 36% tipo 2), tabaquismo diario actual (28%), insuficiencia cardiaca congestiva clase I o II de la Asociación Cardiaca de Nueva York (23%) y eGFR < 60 mL/min por 1.73 m2 (6%). La mayoría de los pacientes tomaban una terapia con estatinas alta (69%) o de intensidad moderada (30%) al inicio del estudio, y el 5% también tomaba ezetimiba. La mayoría de los pacientes tomaban al menos otro medicamento cardiovascular, incluidos los antiagregantes plaquetarios (93%), los bloqueadores beta (76%), los inhibidores de la enzima convertidora de angiotensina (ECA) (56%) o los bloqueadores de los receptores de angiotensina (23%). En la terapia de reducción de lípidos basal estable, la mediana [Q1, Q3] de LDL-C al inicio del estudio fue 92 [80, 109] mg/dL; la media (DE) fue de 98 (28) mg/dL. REPATHA® redujo significativamente el riesgo para la variable primaria compuesta (tiempo hasta la primera aparición de muerte cardiovascular, infarto de miocardio, enfermedad vascular cerebral isquémica, hospitalización por angina inestable o revascularización coronaria; p < 0,0001) y la variable secundaria clave compuesta (tiempo hasta la primera aparición de muerte por enfermedad cardiovascular, infarto de miocardio o enfermedad vascular cerebral isquémica; p < 0,0001). Al realizar el análisis individual de la variable primaria compuesta (tiempo hasta la primera aparición de muerte cardiovascular, infarto de miocardio, enfermedad vascular cerebral isquémica, hospitalización por angina inestable o revascularización coronaria) sólo se demostró beneficio en la reducción del riesgo de infarto de miocardio, enfermedad cerebral vascular isquémica y revascularización coronaria. Las estimaciones de Kaplan Meier de la incidencia acumulada de las variables compuestas primaria y secundaria clave compuesta a lo largo del tiempo. Los resultados de las variables primaria y secundaria se muestran en la Tabla 1 a continuación.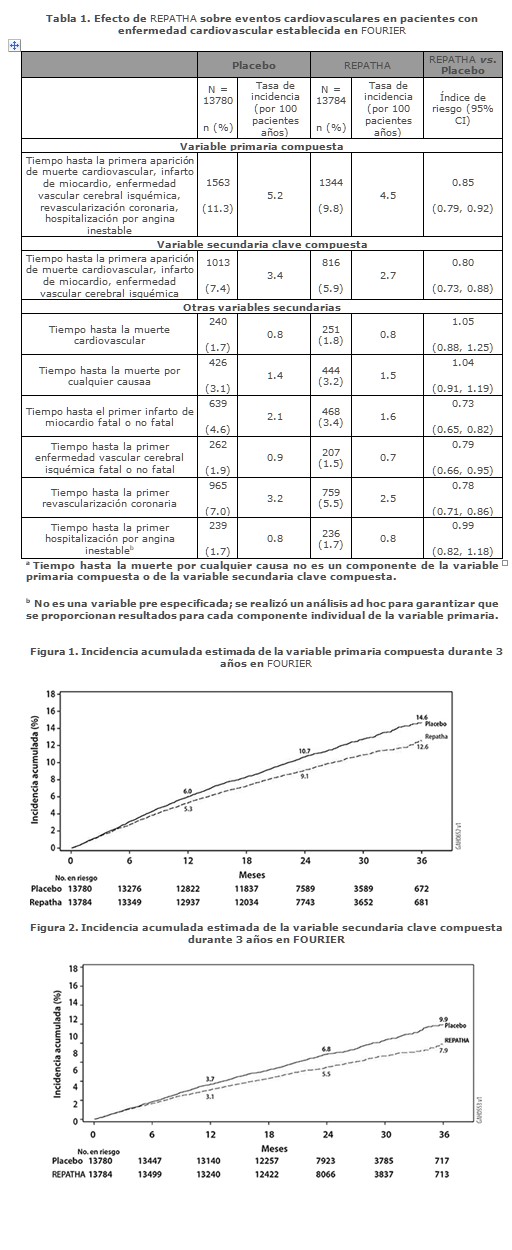
La diferencia entre REPATHA y placebo en el cambio porcentual medio en LDL-C desde la basal hasta la Semana 12 fue de -63% (IC 95%: -63%, -62%) y desde la basal hasta la semana 72 fue -57% (IC 95%: -58%, -56%). En la semana 48, la mediana [Q1, Q3] de LDL-C fue 26 [15, 46] mg/dL en el grupo de REPATHA, con un 47% de pacientes que tenían LDL-C < 25 mg/dL. Teniendo en cuenta todas las evaluaciones, entre los pacientes tratados con REPATHA, 10401 (76%) tenían al menos un valor de LDL-C < 25 mg/dL. Aunque no fue una comparación aleatorizada, el perfil de seguridad fue similar entre pacientes tratados con Repatha con LDL-C después de la basal < 25 mg/dL en comparación con pacientes tratados con Repatha con LDL-C después de la basal (LDL-C ≥ 40 mg/dL). En EBBINGHAUS, un subestudio de 1974 pacientes inscritos en el estudio FOURIER, Repatha no mostró inferioridad comparado con placebo en dominios seleccionados de funciones cognitivas evaluados con el uso de pruebas de función neuropsicológica durante un seguimiento medio de 19 meses. Eficacia Clínica en Hipercolesterolemia Primaria y la Dislipidemia Mixta: Con REPATHA, la reducción de LDL-C de entre el 55% y el 75% aproximadamente se logró incluso en la semana 1 y se mantuvo durante el tratamiento a largo plazo. La respuesta máxima por lo general se obtuvo en el plazo de 1 o 2 semanas tras iniciar el tratamiento con 140 mg cada dos semanas y 420 mg una vez al mes. En el 80% a 85% de todos los pacientes tratados con cualquiera de estas dosis, REPATHA demostró una reducción de LDL-C ≥ 50% en la media de las semanas 10 y 12. Hasta un 99% de los pacientes tratados con cualquiera de las dosis de REPATHA lograron un LDL-C < 1100 mg/dL y hasta un 95% lograron un LDL-C < 70 mg/dL en la media de las semanas 10 y 12. Ambas dosis de REPATHA resultaron eficaces respecto a placebo y ezetimiba en todos los subgrupos, sin diferencias notables entre ellos; los subgrupos estaban determinados por la edad, raza, sexo, región, índice de masa corporal, riesgo según el Programa Nacional de Educación en Colesterol, tabaquismo actual, factores de riesgo de cardiopatía coronaria (CC) basales, antecedentes familiares de CC prematura, tolerancia a la glucosa (es decir, diabetes mellitus de tipo 2, síndrome metabólico o ninguno), hipertensión, dosis e intensidad de estatinas, PCSK9 libre basal, LDL-C basal y TG basal. REPATHA redujo el LDL-C, C-no-HDL, ApoB, TC, Lp(a), VLDL-C, TG, TC/HDL-C y ApoB/ApoA1 y aumentó el HDL-C en pacientes con dislipidemia mixta. REPATHA fue superior a ezetimiba en la reducción de LDL-C, TC, ApoB, C-no-HDL, Lp(a), TC/HDL-C y ApoB/ApoA1. Combinación con Una Estatina y con Una Estatina y Otros Tratamientos Hipolipemiantes: LAPLACE-2 fue un estudio internacional, multicéntrico, doble ciego, aleatorizado y de 12 semanas de duración en el que participaron 1,896 pacientes con hipercolesterolemia primaria o dislipidemia mixta que fueron aleatorizados para recibir REPATHA en combinación con estatinas (rosuvastatina, simvastatina o atorvastatina). REPATHA se comparó con placebo en los grupos de rosuvastatina y simvastatina, y con placebo y ezetimiba en el grupo de atorvastatina. REPATHA redujo significativamente el LDL-C desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo en los grupos de rosuvastatina y simvastatina y en comparación con placebo y ezetimiba en el grupo de atorvastatina (p < 0.001). REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG y Lp(a), y aumentó el HDL-C desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo en los grupos de rosuvastatina y simvastatina (p < 0.05); asimismo, redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1 y Lp(a) en comparación con placebo y ezetimiba en el grupo de atorvastatina (p < 0.001) (ver tablas 2 y 3). RUTHERFORD-2 fue un estudio internacional, multicéntrico, doble ciego, aleatorizado, controlado con placebo y de 12 semanas de duración en el que participaron 329 pacientes con hipercolesterolemia familiar heterocigótica en tratamiento con fármacos hipolipemiantes. REPATHA redujo significativamente el LDL-C desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo (p < 0.001). REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG y Lp(a), y aumentó el HDL-C y ApoA1 desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo (p < 0.05) (ver tabla 2).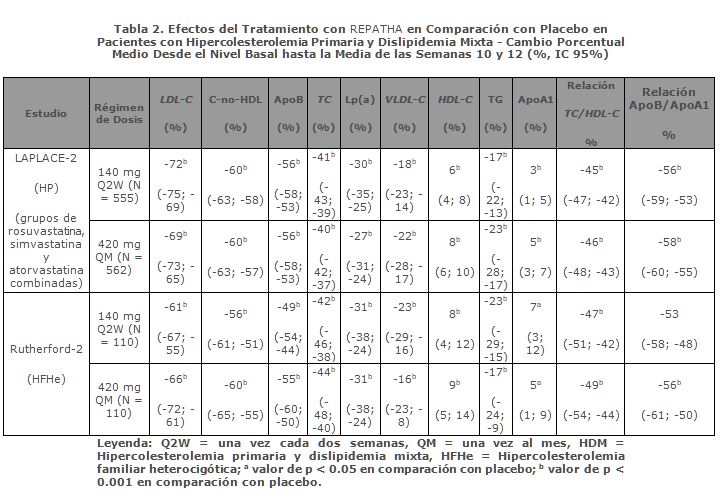
Pacientes Intolerantes a las Estatinas: GAUSS-2 fue un estudio internacional, multicéntrico, doble ciego, aleatorizado, controlado con ezetimiba y de 12 semanas de duración en el que participaron 307 pacientes intolerantes a las estatinas o incapaces de tolerar una dosis eficaz de una estatina. REPATHA redujo significativamente el LDL-C en comparación con ezetimiba (p < 0.001). REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1 y Lp(a) desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con ezetimiba (p < 0.001) (ver tabla 3). Tratamiento en Ausencia de Estatinas: MENDEL-2 fue un estudio internacional, multicéntrico, doble ciego, aleatorizado, controlado con placebo y ezetimiba y de 12 semanas de duración sobre la administración de REPATHA en el que participaron 614 pacientes con hipercolesterolemia primaria y dislipidemia mixta. REPATHA redujo significativamente el LDL-C desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo y ezetimiba (p < 0.001). REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1 y Lp(a) desde el nivel basal hasta la media de las semanas 10 y 12 en comparación con placebo y ezetimiba (p < 0.001) (ver tabla 3).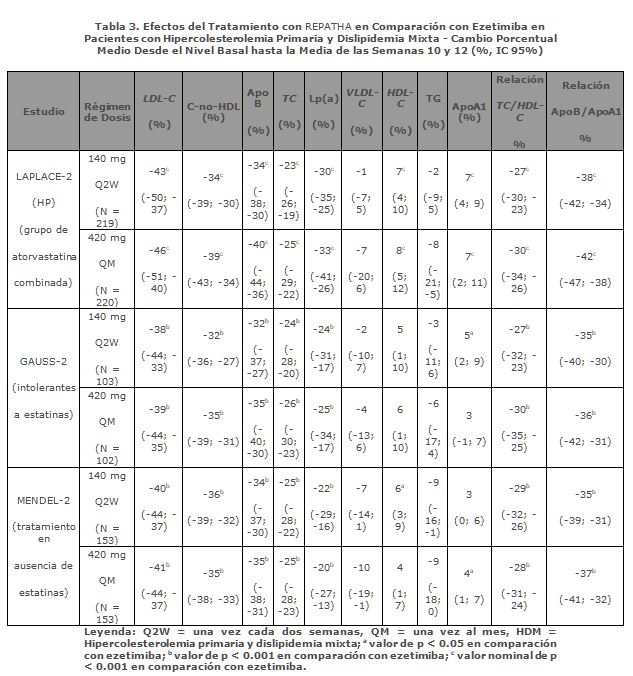
Eficacia a Largo Plazo en la Hipercolesterolemia Primaria y la Dislipidemia Mixta: DESCARTES fue un estudio internacional, multicéntrico, doble ciego, aleatorizado, controlado con placebo y de 52 semanas de duración en el que participaron 901 pacientes con hiperlipidemia controlados únicamente con dieta o bien que seguían un tratamiento con atorvastatina o una combinación de atorvastatina y ezetimiba. REPATHA, en dosis de 420 mg una vez al mes, redujo significativamente el LDL-C respecto al valor basal a las 52 semanas en comparación con placebo (p < 0.001). Los efectos del tratamiento continuaron durante 1 año, como lo demuestra la reducción del LDL-C desde la semana 12 a la 52. La reducción del LDL-C respecto al valor basal en la semana 52 en comparación con placebo no variaba en función del tratamiento hipolipemiante optimizado según los niveles de LDL-C y el riesgo cardiovascular que tenía el paciente. REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG y Lp(a), y aumentó el HDL-C y ApoA1 en la semana 52 en comparación con placebo (p < 0.001) (tabla 4).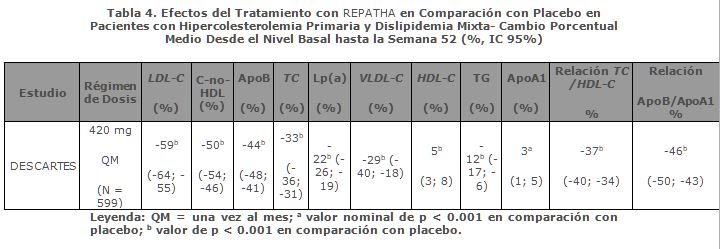
OSLER y OSLER-2 son dos estudios de extensión, abiertos, controlados y aleatorizados actualmente en curso para valorar la seguridad y eficacia a largo plazo de REPATHA en pacientes que finalizaron el tratamiento en un estudio original. En cada estudio de extensión, los pacientes se aleatorizaron 2:1 para recibir REPATHA más el tratamiento estándar (grupo de evolocumab) o únicamente el tratamiento estándar (grupo control) durante el primer año del estudio. Al final del primer año (semana 52 en OSLER y 48 en OSLER-2), los pacientes eran aptos para pasar al periodo de sólo REPATHA en el que todos los pacientes podían recibir REPATHA en abierto durante 4 años más (OSLER) o 1 año más (OSLER-2). En OSLER se incluyeron un total de 1,324 pacientes. REPATHA, en dosis de 420 mg una vez al mes, redujo significativamente el LDL-C respecto al valor basal en la semana 12 y en la semana 52 en comparación con el grupo control (p nominal < 0.001). Los efectos del tratamiento continuaron durante 124 semanas, como lo demuestra la reducción del LDL-C desde la semana 12 del estudio original hasta la semana 112 del estudio de extensión abierto. En OSLER-2 se incluyeron un total de 2,928 pacientes. REPATHA redujo significativamente el LDL-C respecto al valor basal en la semana 12 en comparación con el grupo de control (p nominal < 0.001). Los efectos del tratamiento fueron continuados, como lo demuestra la reducción del LDL-C desde la semana 12 hasta la 24 del estudio de extensión abierto. REPATHA redujo significativamente el TC, ApoB, C-no-HDL, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG y Lp(a), y aumentó el HDL-C y ApoA1 desde el nivel basal hasta la semana 52 en OSLER y la semana 24 en OSLER-2 en comparación con el grupo de control (p nominal < 0.001). El LDL-C y otros parámetros lipídicos volvieron al nivel basal en el plazo de 12 semanas tras la interrupción del tratamiento con REPATHA al comienzo de los estudios OSLER u OSLER-2, sin evidencia de efecto rebote. TAUSSIG es un estudio de extensión, de 5 años de duración, abierto y multicéntrico actualmente en curso para valorar la seguridad y eficacia a largo plazo de REPATHA, como complemento de otros tratamientos hipolipemiantes, en pacientes con hipercolesterolemia familiar grave, incluida la hipercolesterolemia familiar homocigótica. En total, en el estudio TAUSSIG se incluyeron 102 pacientes con hipercolesterolemia familiar grave y 96 con hipercolesterolemia familiar homocigótica. Todos los pacientes del estudio se trataron inicialmente con REPATHA en dosis de 420 mg una vez al mes, excepto los que recibían aféresis en el momento de la inclusión, que empezaron con 420 mg de REPATHA cada dos semanas. La frecuencia de la dosis en los pacientes que no recibían aféresis podía ajustarse hasta 420 mg cada dos semanas en función de la respuesta del LDL-C y los niveles de PCSK9. El uso a largo plazo de REPATHA tuvo un efecto continuado, como lo demuestra la reducción del LDL-C en pacientes con hipercolesterolemia familiar grave (tabla 5). Los cambios en los demás parámetros lipídicos (TC, ApoB, C-no-HDL, TC/HDL-C y ApoB/ApoA1) también demostraron el efecto continuado de la administración a largo plazo de REPATHA en pacientes con hipercolesterolemia familiar grave.
Aún no se ha establecido la relevancia clínica, incluida la seguridad a largo plazo, de los niveles muy reducidos y continuados de LDL-C (es decir, < 25 mg/dL). Los datos disponibles demuestran que no existen diferencias clínicamente significativas entre los perfiles de seguridad de los pacientes con niveles de LDL-C < 0.25 mg/dL y de los pacientes con niveles de LDL-C más altos; ver Reacciones secundarias y adversas. Eficacia Clínica en la Hipercolesterolemia Familiar Homocigótica: TESLA fue un estudio internacional, multicéntrico, doble ciego, aleatorizado, controlado con placebo y de 12 semanas de duración en el que participaron 49 pacientes con hipercolesterolemia familiar homocigótica con edades comprendidas entre los 12 y los 65 años. REPATHA, en dosis de 420 mg una vez al mes, como complemento de otros tratamientos hipolipemiantes (p. ej., estatinas o secuestradores de ácidos biliares), redujo significativamente el LDL-C y ApoB en la semana 12 en comparación con placebo (p < 0.001) (tabla 6). Los cambios observados en los demás parámetros lipídicos (TC, C-no-HDL, TC/HDL-C y ApoB/ApoA1) también demostraron el efecto de la administración de REPATHA en pacientes con hipercolesterolemia familiar homocigótica.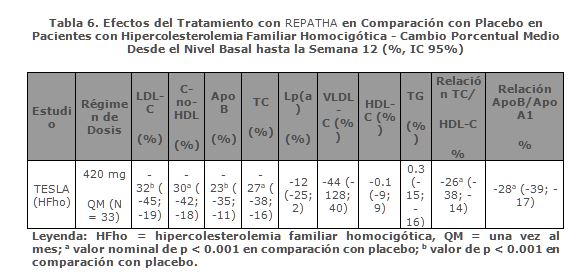
Eficacia a Largo Plazo en la Hipercolesterolemia Familiar Homocigótica: En el estudio TAUSSIG, el uso a largo plazo de REPATHA tuvo un efecto continuado, como lo demuestra la reducción del LDL-C de entre el 20% y el 30% aproximadamente en pacientes con hipercolesterolemia familiar homocigótica no sometidos a aféresis y de entre el 15% y el 25% aproximadamente en pacientes con hipercolesterolemia familiar homocigótica sometidos a aféresis (tabla 7). Los cambios en los demás parámetros lipídicos (TC, ApoB, C-no-HDL, TC/HDL-C y ApoB/ApoA1) también demostraron el efecto continuado de la administración a largo plazo de REPATHA en pacientes con hipercolesterolemia familiar homocigótica. Las reducciones del LDL-C y los cambios en los demás parámetros lipídicos en 13 pacientes adolescentes (≥ 12 a < 18 años) con hipercolesterolemia familiar homocigótica son comparables a los de la población global de pacientes con hipercolesterolemia familiar homocigótica.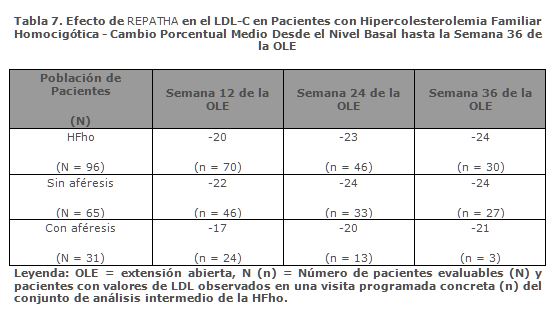
Regresión de la aterosclerosis: GLAGOV fue un estudio de fase 3, doble ciego, aleatorizado, controlado con placebo para evaluar los efectos del tratamiento con REPATHA en la enfermedad aterosclerótica coronaria medida por ultrasonido intravascular (IVUS). Era necesario que los pacientes incluidos en el estudio recibieran terapia hipolipemiante basal estable y que tuvieran un LDL-C ≥ 80 mg /dL o LDL-C ≥ 60 a < 80 mg/dL con un factor de riesgo mayor o tres factores de riesgo cardiovasculares menores. Estos pacientes tenían enfermedad arterial coronaria y requirieron angiografía coronaria. Un total de 970 pacientes fueron aleatorizados 1:1 en dos grupos de tratamiento para recibir REPATHA 420 mg una vez al mes o placebo una vez al mes en inyecciones subcutáneas durante 76 semanas. La ecografía intravascular se realizó al inicio del estudio y en la semana 78. Un total de 27.8% de los pacientes eran mujeres, y 93.8% eran de etnia blanca. La edad media (DE) fue de 59.8 (9.2) años. La media (SD) de LDL-C al inicio fue de 92.6 (27.3) mg/dL. REPATHA redujo el porcentaje de volumen de ateroma (PAV por sus siglas en inglés) y el volumen total de ateroma (TAV por sus siglas en inglés) desde el inicio hasta la semana 78 en comparación con placebo. La regresión de aterosclerosis, definida como cualquier reducción en PAV o TAV en la semana 78, se observó en más pacientes tratados con REPATHA que los pacientes tratados con placebo. Los resultados del estudio se muestran en la tabla 8, a continuación: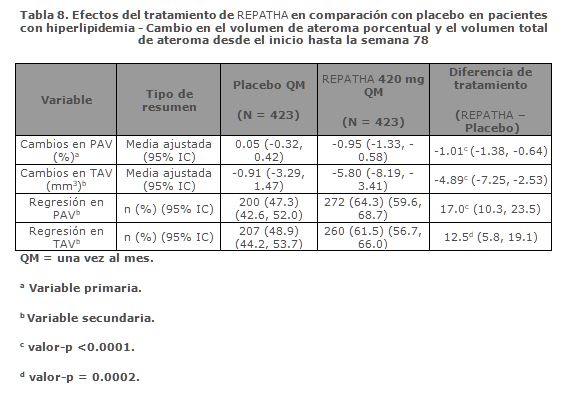
La diferencia de tratamiento en la reducción del LDL-C entre REPATHA y placebo fue del 68.7% (IC del 95%: 64.7%, 72.7%) desde el inicio hasta la semana 78. Estas reducciones se mantuvieron hasta el final del estudio. Las concentraciones medias correspondientes de LDL-C (DE) en la semana 78 fueron 29.2 (27.6) mg/dL en el grupo de REPATHA. Con base en un análisis ad-hoc, las concentraciones más bajas de LDL-C logradas durante el estudio se asociaron con una mayor regresión de la aterosclerosis, medida por la reducción en PAV. Población Pediátrica: Los datos disponibles sobre el uso de REPATHA en la población pediátrica son limitados. En los ensayos clínicos se incluyeron catorce pacientes adolescentes con edades ≥ 12 a < 18 que sufrían hipercolesterolemia familiar homocigótica. No se han observado diferencias generales en la seguridad ni la eficacia entre los pacientes adolescentes y los pacientes adultos con hipercolesterolemia familiar homocigótica. Ver Dosis y vía de administración para información de uso pediátrico.
Contraindicaciones: Hipersensibilidad al principio activo o a los componentes de la fórmula. Hipersensibilidad al látex. No se use en el embarazo, lactancia ni en niños menores de 12 años.
Precauciones generales: Insuficiencia Hepática: En pacientes con insuficiencia hepática moderada se observó una reducción en la exposición total a evolocumab que puede dar lugar a una disminución del efecto sobre la reducción de los niveles de LDL-C. Por lo tanto, en estos pacientes se debe garantizar un estrecho seguimiento.Los pacientes con insuficiencia hepática grave (Child-Pugh clase C) no han sido estudiados (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis y sobre la fertilidad). REPATHA se debe usar con precaución en pacientes con insuficiencia hepática grave. Caucho Natural: La cubierta de la aguja de la jeringa de vidrio prellenada y de la pluma precargada está fabricada con caucho natural (un derivado del látex), que puede causar reacciones alérgicas.Contenido de Sodio: Este medicamento contiene menos de 1 mmol (23 mg) de sodio por dosis, por lo que se considera esencialmente "libre de sodio".
Restricciones de uso durante el embarazo y la lactancia: Embarazo: No hay datos o éstos son limitados relacionados con el uso de REPATHA en mujeres embarazadas. Los estudios en animales no sugieren efectos directos ni indirectos en términos de toxicidad para la reproducción (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis y sobre la fertilidad). REPATHA no debe utilizarse durante el embarazo. Lactancia: Se desconoce si evolocumab se excreta en la leche materna. No se puede excluir el riesgo en recién nacidos/niños alimentados mediante lactancia materna. Se debe decidir si es necesario interrumpir la lactancia materna o interrumpir/no iniciar el tratamiento con REPATHA tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre. Fertilidad: No hay datos disponibles sobre el efecto de evolocumab en la fertilidad humana. Los estudios en animales no mostraron efecto alguno sobre las variables de fertilidad en el área bajo la curva de concentración-tiempo (ABC) de niveles de exposición mucho más altos que en pacientes que reciben evolocumab en dosis de 420 mg una vez al mes (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis y sobre la fertilidad).
Reacciones secundarias y adversas: Resumen del Perfil de Seguridad: Las reacciones adversas del medicamento notificadas con mayor frecuencia durante los estudios pivotales a las dosis recomendadas, fueron nasofaringitis (7.4%), infección respiratoria del tracto superior (4.6%), dolor de espalda (4.4%), artralgia (3.9%), gripe (3.2%), y reacciones en el sitio de inyección (2.2%). El perfil de seguridad de la población con hipercolesterolemia familiar homocigótica fue consistente con lo demostrado en la población con hipercolesterolemia primaria y dislipidemia mixta. Resumen Tabulado de Reacciones Adversas: Las reacciones adversas notificadas en los estudios clínicos pivotales controlados en pacientes con hipercolesterolemia primaria y dislipidemia mixta, enfermedad cardiovascular establecida e hipercolesterolemia familiar homocigótica, y reportes espontáneos se muestran en la tabla 9, la cual aparece a continuación, según el sistema de clasificación de órganos y frecuencia, utilizando la siguiente convención: muy frecuente (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1,000 a < 1/100), raras (≥ 1/10,000 a < 1/1,000) y muy raras ( < 1/10,000).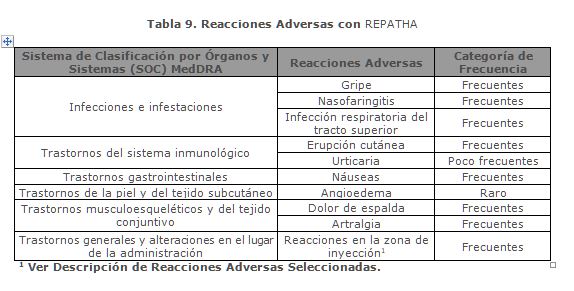
Descripción de Reacciones Adversas Seleccionadas: Reacciones en la Zona de Inyección: Las reacciones más frecuentes en el lugar de la inyección fueron eritema, dolor y hematoma en la zona de aplicación.Población Pediátrica: La experiencia con REPATHA en pacientes pediátricos es limitada. En los estudios clínicos se incluyeron catorce pacientes con edades entre ≥ 12 a < 18 que presentaban hipercolesterolemia familiar homocigótica. No se observó ninguna diferencia en la seguridad entre los adolescentes y los pacientes adultos con hipercolesterolemia familiar homocigota. No se ha establecido la seguridad y eficacia de REPATHA en pacientes pediátricos con hipercolesterolemia primaria y dislipidemia mixta. Población de Edad Avanzada: Aunque no se observaron problemas de seguridad en los pacientes mayores de 75 años, los datos son limitados en este subgrupo de edad. De los 6,026 pacientes totales incluidos en los estudios clínicos de REPATHA, 1,779 (30%) tuvieron ≥ 65 años y 223 (4%) ≥ 75 años. En general, no se observaron diferencias en la seguridad ni la eficacia entre estos pacientes y los pacientes más jóvenes.Inmunogenicidad: En un grupo de ensayos clínicos controlados activos y con placebo, el 0,3% (48 de 17,992) de los pacientes tratados con al menos una dosis de REPATHA dieron positivo para el desarrollo de anticuerpos de unión. Los pacientes cuyos sueros dieron positivo para anticuerpos de unión se evaluaron adicionalmente para anticuerpos neutralizantes; ninguno de los pacientes dio positivo para anticuerpos neutralizantes. No hubo pruebas de que la presencia de anticuerpos antiadherentes afectara el perfil farmacocinético, la respuesta clínica o la seguridad de REPATHA, pero se desconocen las consecuencias a largo plazo del tratamiento con REPATHA en presencia de anticuerpos que se unen a fármacos.
Interacciones medicamentosas y de otro género: No se han realizado estudios formales de interacción fármaco-fármaco para REPATHA. La interacción farmacocinética entre las estatinas y evolocumab fue evaluada en los estudios clínicos de REPATHA. Se observó un incremento aproximado de 20% en la depuración de evolocumab en pacientes co-administrados con estatinas. Este aumento en la depuración es en parte mediado por el efecto de las estatinas que incrementan la concentración de PCSK9, hecho que no impactó adversamente el efecto de farmacodinamia de REPATHA sobre los lípidos. No se requiere ajuste a la dosis de la estatina cuando se usa en combinación con REPATHA. No se han realizado estudios de interacciones farmacocinéticas ni farmacodinámicas entre REPATHA y fármacos hipolipemiantes distintos de las estatinas y la ezetimiba. En ausencia de estudios de compatibilidad, este medicamento no debe ser mezclado con otros productos medicinales.
Alteraciones en los resultados de pruebas de laboratorio: Ninguna conocida.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Evolocumab no fue carcinogénico en hámsters con exposiciones muy superiores a las de los pacientes que recibían evolocumab a dosis de 420 mg una vez al mes. No se ha evaluado el potencial mutagénico de evolocumab. En hámsters y monos macacos con exposiciones muy superiores a las de los pacientes que recibían evolocumab a dosis de 420 mg una vez al mes, no se observaron efectos sobre la fertilidad de los machos ni de las hembras. En monos macacos con exposiciones muy superiores a las de los pacientes que recibían evolocumab en dosis de 420 mg una vez al mes, no se observaron efectos sobre el desarrollo embriofetal ni posnatal (hasta los 6 meses de edad). A excepción de una Respuesta Dependiente de Anticuerpos de las células T en monos macacos inmunizados con KLH después de 3 meses de tratamiento con evolocumab, no se observaron efectos adversos en hámsters (hasta los 3 meses) ni en monos macacos (hasta los 6 meses) con exposiciones muy superiores a las de los pacientes que recibían evolocumab en dosis de 420 mg una vez al mes. En estos estudios, el efecto farmacológico pretendido sobre la disminución del LDL-C y colesterol total en suero fue observado y fue reversible tras la interrupción del tratamiento. No se observaron efectos adversos en combinación con rosuvastatina durante 3 meses en monos macacos con exposiciones muy superiores a las de los pacientes que recibían 420 mg de evolocumab una vez al mes. Las reducciones del LDL-C y colesterol total en suero fueron más pronunciadas que las observadas anteriormente con evolocumab solo, y reversibles tras la interrupción del tratamiento.
Dosis y vía de administración: Antes de iniciar el tratamiento con REPATHA, se deben excluir las causas secundarias de la hipercolesterolemia o dislipidemia mixta (p. ej., el síndrome nefrótico y el hipotiroidismo). Posología: Enfermedad Cardiovascular Establecida e Hipercolesterolemia Primaria y Dislipidemia Mixta en Adultos: La dosis recomendada de REPATHA es 140 mg cada 2 semanas o 420 mg una vez al mes; ambas dosis son clínicamente equivalentes. Hipercolesterolemia Familiar Homocigota en Adultos y Adolescentes de 12 Años de Edad y Mayores: La dosis inicial recomendada es 420 mg una vez al mes. Después de 12 semanas de tratamiento, la frecuencia de la dosificación se puede ajustar al alza a 420 mg cada dos semanas si no se obtiene una respuesta clínicamente significativa. Los pacientes en aféresis pueden iniciar el tratamiento con 420 mg cada 2 semanas para que coincida con su programa de aféresis. Pacientes con Insuficiencia Renal: No se necesita ajuste de la dosis en pacientes con insuficiencia renal (vea Farmacocinética y farmacodinamia [poblaciones especiales]). Pacientes con Insuficiencia Hepática: No se necesita ajuste de la dosis en pacientes con insuficiencia hepática leve, ver Precauciones generales para pacientes con insuficiencia hepática moderada y severa. Pacientes Geriátricos (≥ 65 Años de Edad): No se necesita ajuste de la dosis en pacientes geriátricos. Población Pediátrica: No se ha establecido la seguridad y eficacia de REPATHA en niños menores de 18 años en la indicación para hipercolesterolemia primaria y dislipidemia mixta. No se dispone de datos. Por lo anterior no deberá emplearse en esta población. No se ha establecido la seguridad y eficacia de REPATHA en niños menores de 12 años en la indicación para hipercolesterolemia familiar homocigota. No se dispone de datos. Por lo anterior no deberá emplearse en esta población. Método de Administración: Vía subcutánea. REPATHA se debe administrar mediante inyección subcutánea en el abdomen, el muslo o la parte superior del brazo. Se deben alternar las zonas de inyección y descartar aquéllas donde la piel presente dolor a la palpación, equimosis, eritema o induración. REPATHA no se debe administrar por vía intravenosa ni intramuscular. La dosis de 420 mg una vez al mes o cada dos semanas se debe administrar utilizando tres jeringas prellenadas o tres plumas precargadas administradas consecutivamente dentro de un intervalo de 30 minutos. REPATHA está pensado para que el paciente se lo autoadministre después de recibir una capacitación adecuada. La administración de REPATHA también puede realizarla una persona que haya sido capacitada para administrar el medicamento. Cada jeringa prellenada o pluma precargada es de un solo uso. Para consultar las instrucciones de administración, vea el instructivo. Precauciones Especiales de Eliminación y Otras Manipulaciones: La