ROACTEMRA
ROCHE
Denominación genérica: Tocilizumab.
Forma farmacéutica y formulación: Solución. El frasco ámpula contiene: tocilizumab 80 mg, 200 mg y 400 mg. Vehículo cbp 4, 10 y 20 ml.
Indicaciones terapéuticas: ROACTEMRA® está indicado para el tratamiento de la artritis reumatoide (AR) activa de intensidad moderada a grave, en pacientes adultos. ROACTEMRA® se puede utilizar solo o en combinación con metotrexato (MTX) y/u otros fármacos antireumáticos modificadores de la enfermedad (FARME).
Farmacocinética y farmacodinamia: En los estudios clínicos con tocilizumab, se observó una rápida disminución en los niveles de proteína C reactiva (PCR), en la velocidad de sedimentación globular (VSG) y en la proteína amiloide A sérica. Se observó incremento en los niveles de hemoglobina, debido a que tocilizumab disminuye los efectos producidos por IL-6 sobre la producción de hepcidina y de esta manera incrementa la disponibilidad del hierro. Mecanismo de acción: ROACTEMRA® es un anticuerpo monoclonal dirigido contra el receptor de la interleucina-6 (IL-6R) antihumana, humanizada recombinante de la subclase de inmunoglobulina (Ig) IgG1. ROACTEMRA® se une específicamente a los receptores de IL-6 tanto solubles como los unidos a la membrana (sIL-6R y mIL-6R), y ha demostrado que inhibe la cascada de señalización mediada por sIL-6R y mIL-6R. La IL-6 es una citocina multifuncional, producida por una gran variedad de tipos de células involucradas en la función paracrina local así como la regulación de los procesos fisiológicos y patológicos sistémicos tales como la inducción de la secreción de inmunoglobulinas, la activación de linfocitos T, la inducción de proteínas hepáticas de fase aguda y la estimulación de hematopoyesis. La IL-6 ha sido implicada en la patogénesis de varias enfermedades, incluyendo las enfermedades inflamatorias, la osteoporosis y neoplasias. Eficacia clínica: la eficacia de ROACTEMRA® en el alivio de los signos y síntomas de la artritis reumatoide fue evaluada en cinco estudios multicéntricos, doble ciego y aleatorizados. Como criterios de inclusión a dichos estudios requirieron pacientes ≥18 años de edad con artritis reumatoide activa, diagnosticados de acuerdo con los criterios del Colegio Americano de Reumatología (ACR por sus siglas en inglés), quienes deberían presentar por lo menos 8 articulaciones dolorosas y 6 articulaciones inflamadas en el registro basal. ROACTEMRA® fue administrado intravenosamente cada 4 semanas como monoterapia (Estudio I), en combinación con MTX (Estudios II, III, V) o con otros FARME (Estudio IV). El Estudio I evaluó a 673 pacientes quienes no habían sido tratados con MTX dentro de los 6 meses previos a la aleatorización; en pacientes que habían recibido tratamiento con MTX antes de dicho periodo, el motivo de la suspensión del tratamiento debió ser por una causa distinta a toxicidad o ineficacia(falta de respuesta). La mayoría (67%) de los pacientes eran vírgenes al tratamiento con MTX. Tocilizumab se administró a una dosis de 8 mg/kg cada cuatro semanas como monoterapia. Al grupo control se le administró semanalmente MTX (dosis escalonada de 7,5 mg hasta un máximo de 20 mg, en un período de 8 semanas). La medida de desenlace primario fue la proporción de pacientes quienes alcanzaron una respuesta ACR20 en la semana 24. El Estudio II, un estudio de 2 años de duración que se está llevando a cabo, con un análisis preliminar planeado en la semana 24, evaluó a 1.196 pacientes quienes habían tenido una respuesta clínica inadecuada a MTX. Los pacientes se agruparon de manera aleatoria para recibir tocilizumab a dosis de 4 u 8 mg/kg o placebo cada cuatro semanas de manera cegada durante 52 semanas, en combinación con MTX con dosis estable (10-25 mg semanalmente). La medida de desenlace primario en la semana 24 fue la proporción de pacientes quienes alcanzaron respuesta ACR20. El Estudio III evaluó a 623 pacientes quienes habían tenido una respuesta clínica inadecuada a MTX. Las dosis de 4 u 8 mg/kg de tocilizumab ó placebo fueron administradas cada 4 semanas, en combinación con dosis estable de MTX (10-25 mg semanalmente). El Estudio IV evaluó a 1.220 pacientes quienes tuvieron una respuesta inadecuada a la terapia convencional, incluyendo uno o más FARME. Las dosis de 8 mg/kg de tocilizumab o placebo se administraban cada cuatro semanas, en combinación con la terapia estable con FARME. El Estudio V evaluó a 499 pacientes quienes tuvieron una respuesta clínica inadecuada o fueron intolerantes a una o más terapias antiTNF. El agente antiTNF fue suspendido antes de la aleatorización. Las dosis de 4 u 8 mg/kg de tocilizumab o placebo fueron administradas cada cuatro semanas, en combinación con MTX con dosis estable (10-25 mg semanalmente). La medida de desenlace primario para los estudios III-V fue la proporción de pacientes quienes alcanzaron una respuesta ACR20 en la semana 24. El porcentaje de pacientes quienes lograron respuestas ACR 20, 50 y 70 en los Estudios I a V se muestran en la tabla 1.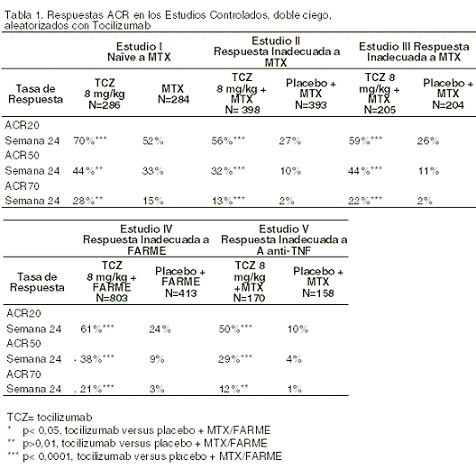
En todos los estudios, los pacientes tratados con tocilizumab tuvieron tasas de respuesta de ACR20, 50, 70, más elevadas, estadísticamente significativas, a los 6 meses, en comparación al control. El efecto del tratamiento fue similar en los pacientes independientemente del estado del factor reumatoide, la edad, el sexo, la raza, el número de tratamientos previos ó el estado de la enfermedad. La respuesta al tratamiento con tocilizumab fue rápida (tan temprano como a la semana 2) y la magnitud de la respuesta mejoró durante el tratamiento. Se observaron respuestas durables continuas durante más de 18 meses en los estudios abiertos de extensión que se están llevando a cabo, de los Estudio I, III-V. En los pacientes tratados con tocilizumab, se observaron mejorías significativas en todos los componentes individuales de la respuesta ACR (articulaciones dolorosas e inflamadas, evaluación global de la enfermedad por parte de los pacientes y del médico, calificaciones de los índices de discapacidad (HAQ), evaluación del dolor y PCR, en comparación a los pacientes que estaban recibiendo placebo + MTX /FARME en todos los estudios. Los pacientes tratados con tocilizumab tuvieron una reducción significativamente mayor en la calificación de la actividad de la enfermedad (DAS28) en comparación a los pacientes tratados con placebo + FARME. Una respuesta EULAR (European League Against Rheumatism) buena a moderada se logró en un número significativamente mayor de pacientes tratados con tocilizumab, en comparación a los pacientes tratados con placebo + FARME (ver tabla 2).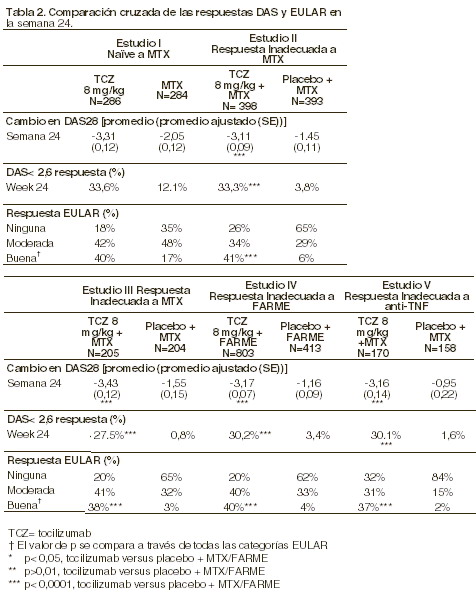
Resultados de la calidad de vida: se observaron mejorías clínicamente significativas en el índice de discapacidad (HAQ-DI, Indice de Discapacidad del Cuestionario de Evaluación de la Salud), fatiga (FACIT-F, Evaluación Funcional del Tratamiento en Enfermedad Crónica-Fatiga) y mejoría en la salud física SF-36 (Forma Corta 36), en los componentes físico y mental, en los pacientes tratados con tocilizumab (monoterapia o combinación con FARME), en comparación a los pacientes tratados con MTX/FARME (ver tabla 3). En la semana 24, la proporción de pacientes tratados con tocilizumab que mostraban una mejoría clínicamente relevante en el HAQ-DI (definido como una disminución > 0,25 en la calificación total individual), fue significativamente más elevada en comparación a los pacientes que estaban recibiendo placebo + MTX/FARME en todos los estudios.
Evaluaciones de laboratorio: el tratamiento con ROACTEMRA® como monoterapia o en combinación con FARME /MTX resultó en una elevada mejoría estadísticamente significativa en los niveles de hemoglobina, en comparación con placebo + MTX/ FARME (p < 0,0001) en la semana 24. La mejoría más importante se observó en los pacientes con anemia crónica asociada con AR; los niveles promedio de hemoglobina se incrementaron en la semana 2 y permanecieron dentro del rango normal hasta la semana 24. Una marcada disminución en los niveles promedio de PCR, VSG y proteína amiloide A sérica ocurrió rápidamente después de la administración de tocilizumab. Consistente con el efecto sobre los reactantes de fase aguda, el tratamiento con tocilizumab se asoció con una reducción en la cuenta de plaquetas hasta un rango normal. Farmacocinética: la farmacocinética de ROACTEMRA® fue determinada utilizando una población para el análisis de farmacocinética en una base de datos compuesta de 1.793 pacientes con artritis reumatoide tratados con una infusión durante una hora, de 4 y 8 mg/kg, cada 4 semanas durante 24 semanas. Los parámetros de farmacocinética de ROACTEMRA® no cambian con el tiempo. Se observó un incremento proporcional a la dosis en el área bajo la curva (ABC) y se observó una concentración sostenida (Cmín) para las dosis de 4 y 8 mg/kg, cada 4 semanas. La concentración máxima (Cmáx) se incrementó en forma proporcional a la dosis. En estado estable, la ABC pronosticada y la Cmín fueron 2,7 y 6,5 veces más elevadas con la dosis de 8 mg/kg en comparación a 4 mg/kg, respectivamente. Los siguientes parámetros son válidos para una dosis de 8 mg/kg de tocilizumab administrada cada 4 semanas. El promedio pronosticado (± DE) de la ABC en estado estable, la Cmín y la Cmáx de tocilizumab fueron de 35 000 ± 15 500 h•mg/ml, 9,74 ± 10,5 mg/ml y 183 ± 85,6mg/ml, respectivamente. Las relaciones de acumulación para la ABC y la Cmáx fueron pequeñas; 1,22 y 1,06, respectivamente. La relación de la acumulación fue más elevada para la Cmín (2,35), la cual era esperada en base a la contribución de la depuración no lineal en concentraciones más bajas. El estado estable se alcanzó después de la primera administración y después de 8 y 20 semanas para la Cmáx, la ABC y la Cmín, respectivamente. Distribución: después de la administración IV, tocilizumab es sometido a una eliminación bifásica de la circulación. En los pacientes con artritis reumatoide, el volumen de distribución central fue de 3,5 l, el volumen de distribución periférico fue de 2,9 l, resultando en un volumen de distribución en estado estable de 6,4 l (22) Eliminación: la depuración total de tocilizumab fue dependiente de la concentración y es la suma de la depuración lineal y la depuración no lineal. La depuración lineal se estimó como un parámetro en la población para el análisis de farmacocinética y fue de 12,5 ml/h. La depuración no lineal dependiente de la concentración juega un papel importante en las concentraciones bajas de tocilizumab. Una vez que la vía de depuración no lineal se satura, en las concentraciones elevadas de tocilizumab, la depuración se determina principalmente por medio de la depuración lineal. La t½ de tocilizumab depende de la concentración. En el estado estable después de la administración de una dosis de 8 mg/kg cada 4 semanas, la t½ disminuyó al disminuir las concentraciones dentro de un intervalo de dosificación de 14 a 8 días. Farmacocinética en poblaciones especiales: insuficiencia hepática: no se ha realizado ningún estudio formal del efecto de la insuficiencia hepática sobre la farmacocinética de tocilizumab. Insuficiencia renal: no se ha realizado ningún estudio formal del efecto de la insuficiencia renal sobre la farmacocinética de tocilizumab. La mayoría de los pacientes en la población para el análisis de farmacocinética tuvieron una función renal normal o insuficiencia renal leve. La insuficiencia renal leve (depuración de creatinina basada en Cockcroft -Gault < 80 ml/min y > 50 ml/min) no impactó la farmacocinética de tocilizumab. Otras poblaciones especiales: los análisis de población evaluaron los efectos potenciales de las características demográficas sobre la farmacocinética de tocilizumab en los pacientes adultos con artritis reumatoide. Los resultados de esos análisis mostraron que no es necesario un ajuste en la dosis debido a la edad, sexo o raza.
Contraindicaciones: Hipersensibilidad a los componentes del fármaco. El tratamiento con ROACTEMRA® no debe ser iniciado en pacientes con infecciones activas graves.
Precauciones generales: Infecciones: el tratamiento con ROACTEMRA® no se debe iniciar en los pacientes con infecciones activas graves. La administración de ROACTEMRA® deberá interrumpirse si un paciente desarrolla una infección grave, hasta que la infección sea controlada. Los médicos deben tener precaución cuando consideren utilizar ROACTEMRA® en los pacientes con historia de infección recurrente o condiciones subyacentes (por ejemplo diverticulitis, diabetes), lo cual puede predisponer a los pacientes a las infecciones. Se recomienda la vigilancia para la detección oportuna de una infección grave en los pacientes que están recibiendo tratamientos biológicos para la artritis reumatoide de intensidad moderada a grave, debido a que los signos y síntomas de la inflamación aguda pueden disminuirse, asociados con la supresión de la reacción de la fase aguda. A los pacientes se les debe indicar que deben contactar al médico inmediatamente cuando aparezca cualquier síntoma sugerente de infección, con el objeto de asegurar la rápida evaluación y el tratamiento apropiado. Las vacunas vivas y atenuadas no se deberán administrar concurrentemente con ROACTEMRA®, debido a que no se ha establecido la seguridad clínica. No existen datos disponibles sobre la transmisión secundaria de la infección por las personas que están recibiendo vacunas vivas, a los pacientes que están recibiendo ROACTEMRA®. El tratamiento con ROACTEMRA® debe ser interrumpido si los pacientes desarrollan infecciones serias hasta que la infección sea controlada. Reacciones de hipersensibilidad: se han reportado reacciones de hipersensibilidad graves, en asociación con la infusión de ROACTEMRA®en el 0,3% de los pacientes (ver Reacciones secundarias y adversas). El tratamiento apropiado debe estar disponible para el uso inmediato en el caso de una reacción anafiláctica durante la administración de ROACTEMRA®. Enfermedad hepática activa e insuficiencia hepática: el tratamiento con ROACTEMRA®, particularmente cuando se administra concomitantemente con metotrexato, se puede asociar con incrementos en los niveles de transaminasas hepáticas (ver Alteraciones en los resultados de pruebas de laboratorio). Por lo tanto, se debe tener precaución al considerar el tratamiento de los pacientes con enfermedad hepática activa o insuficiencia hepática (ver Instrucciones para dosis especiales). Abuso y dependencia de fármacos: no se han realizado estudios sobre los efectos en el potencial de tocilizumab para producir dependencia. Sin embargo, no existe evidencia de los datos disponibles de que el tratamiento de ROACTEMRA® resulta en dependencia. Capacidad para conducir y uso de maquinaria: no se han realizado estudios sobre la capacidad para conducir y el uso de maquinaria. Sin embargo, no existe evidencia a partir de los datos disponibles, de que el tratamiento con ROACTEMRA® afecta la capacidad para conducir y el uso de máquinas. Neutropenia: se debe tener precaución al iniciar el tratamiento de tocilizumab en los pacientes con una baja cuenta de neutrófilos. Las disminuciones en la cuenta de neutrófilos por debajo de 1 x 109/l se presentaron en el 3,4% de los pacientes, con cuentas < 0,5 x 109/l, en el 0,3% de los pacientes que estaban recibiendo 8 mg/kg + FARME sin una clara asociación con la infección grave (ver Alteración en los resultados de pruebas de laboratorio). En los pacientes con una cuenta absoluta de neutrófilos < 0,5 x 109/l, no se recomienda el tratamiento. Inmunocompromiso: el tratamiento con ROACTEMRA® no deberá ser iniciado si el paciente presenta un sistema inmunológico deficiente. Diabetes mellitus: en los estudios clínicos de desarrollo de ROACTEMRA®, se realizó un análisis de seguridad en poblaciones especiales con la intención de evaluar la incidencia de eventos adversos en dichas poblaciones. La población de pacientes con diabetes mellitus que ingresó a los estudios correspondió a 8,4% de la muestra total. El análisis de los datos hasta marzo 2008 demostró que en todos los grupos de tratamiento (ROACTEMRA® o con placebo), los pacientes con diabetes mellitus tuvieron una tasa mayor de infecciones (38,6%, grupo placebo vs. 43,5% grupo con ROACTEMRA®), comparado con los pacientes no diabéticos (30,8%, grupo placebo vs. 36,2 grupo con ROACTEMRA®). El patrón de las infecciones no fue diferente del observado en otras poblaciones, siendo las infecciones de vías respiratorias altas el evento adverso más frecuente. La incidencia de eventos adversos serios en pacientes con diabetes mellitus fue similar en los grupos de pacientes que recibieron ROACTEMRA® y en aquellos que recibieron placebo (10,2% y 10,9%, respectivamente). La infección seria más común que ocurrió en ambos grupos de pacientes fueron neumonía y celulitis. Dentro del análisis de seguridad en la población de pacientes con diabetes mellitus no hubo evidencia de que existiera correlación con cambios en otros aspectos importantes de esta enfermedad como en los niveles de creatinina sérica, niveles de presión arterial e incremento del peso corporal en comparación con la población de pacientes no diabéticos. Debido al incremento en las infecciones serias reportadas con todas las terapias biológicas para artritis reumatoide, los médicos prescriptores y al cuidado de los pacientes deberán informar a los pacientes sobre este riesgo y trabajar en conjunto con ellos para el manejo del riesgo, vigilancia y detección temprana de una infección clínica. Uso pediátrico, uso geriátrico y uso en insuficiencia renal: los análisis de población evaluaron los efectos potenciales de las características demográficas sobre la farmacocinética de tocilizumab en los pacientes adultos con artritis reumatoide. Los resultados de esos análisis mostraron que no es necesario un ajuste en la dosis debido a la edad, sexo o raza. Tuberculosis: ROACTEMRA®no debe ser usado en pacientes con tuberculosis activa. Antes de iniciar el tratamiento con ROACTEMRA® todos los pacientes deben ser evaluados para identificar factores de riesgo de tuberculosis y signos y síntomas de tuberculosis activa mediante un examen clínico minucioso. Las normas locales deben ser aplicadas para la evaluación de pacientes con tuberculosis latente y para su tratamiento si las pruebas resultan positivas. En pacientes con pruebas cutáneas positivas y radiografía de tórax negativa, el tratamiento con ROACTEMRA® puede ser iniciado un mes después de iniciada la terapia antituberculosa. No se ha realizado ningún estudio formal del efecto de la insuficiencia renal sobre la farmacocinética de tocilizumab. La mayoría de los pacientes en la población para el análisis de farmacocinética tuvieron una función renal normal o insuficiencia renal leve. La insuficiencia renal leve (depuración de creatinina basada en Cockcroft -Gault < 80 ml/min y > 50 ml/min) no impactó la farmacocinética de tocilizumab.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no existen datos adecuados del uso de tocilizumab en las mujeres embarazadas. Un estudio realizado en animales ha demostrado un incremento en el riesgo de aborto espontáneo/fallecimiento embrio-fetal con la dosis elevada. Se desconoce el potencial riesgo para los humanos. ROACTEMRA® no deberá utilizarse durante el embarazo a menos que sea claramente necesario. Lactancia: no se sabe si ROACTEMRA® es excretado en la leche humana. Aun cuando las inmunoglobulinas endógenas del isotipo IgG son secretadas en la leche humana, es improbable una absorción sistémica de ROACTEMRA® por medio del amamantamiento debido a una rápida degradación proteolítica de dichas proteínas en el sistema digestivo. La decisión de continuar o suspender el amamantamiento o continuar/suspender la terapia con ROACTEMRA®deberá realizarse tomando en consideración el beneficio de la lactancia para el bebé y el beneficio de la terapia con ROACTEMRA® para la mujer.
Reacciones secundarias y adversas: Estudios clínicos: un total de 3.728 pacientes recibieron por lo menos una dosis de ROACTEMRA®. Las reacciones adversas producidas por el fármaco (ReAd) presentadas en la siguiente tabla, se basan en la seguridad de ROACTEMRA®, y fueron evaluadas en 4 estudios controlados con placebo y en 1 estudio controlado con MTX. En esos estudios, 774 pacientes recibieron dosis de 4 mg/kg de ROACTEMRA® en combinación con MTX, 1.582 pacientes recibieron dosis de 8 mg/kg de ROACTEMRA® en combinación con MTX /otros FARME y 288 pacientes recibieron monoterapia con 8 mg/kg de ROACTEMRA®. En los estudios a largo plazo, abiertos y de extensión a, se incluyeron a 2.439 pacientes quienes recibieron dosis de 8 mg/kg de ROACTEMRA® con y sin FARME. La exposición total en el análisis de seguridad a largo plazo fue de 2.628 pacientes/años. Las reacciones adversas producidas por el fármaco se enlistan de acuerdo a la importancia clínica. Las frecuencias se definen como muy comunes ≥ 1/10, comunes ≥ 1/100 a < 1/10 o no comunes ≥ 1/1.000 a < 1/100.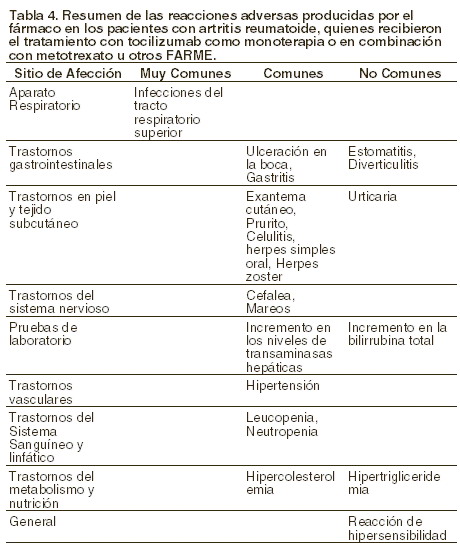
Infecciones: en los estudios controlados, la tasa de todas las infecciones reportadas con la dosis de 8 mg/kg de ROACTEMRA® + FARME fue de 118 eventos por cada 100 pacientes (pt) años, en comparación a 104 eventos por 100 pt años en el grupo tratado con placebo + FARME. En los estudios abiertos y de extensión, a largo plazo, la tasa de infecciones con tocilizumab + FARME fue de 113 eventos por 100 pacientes años de exposición. En los estudios clínicos controlados, la tasa de infecciones graves con dosis de 8 mg/kg de ROACTEMRA® + FARME fue de 5,2 eventos por 100 pacientes años de exposición, en comparación a 3,8 eventos por 100 pacientes años de exposición en el grupo tratado con placebo + FARME. En el estudio con monoterapia, la tasa de infecciones graves fue de 2,9 eventos por 100 pacientes años de exposición en el grupo tratado con ROACTEMRA® y 1,5 eventos por 100 pacientes años de exposición en el grupo tratado con MTX. En la población para el estudio de seguridad a largo plazo (estudios centrales y de extensión), la tasa de infecciones graves observadas con el tratamiento de ROACTEMRA® + FARME fue de 3,8 eventos por 100 pacientes años de exposición. Las infecciones graves reportadas incluyeron neumonía, celulitis, herpes zóster, gastroenteritis, diverticulitis, sepsis, artritis bacteriana. Rara vez, las infecciones graves fueron fatales. Se reportaron casos únicos de infecciones por oportunistas, las cuales respondieron al tratamiento: por ejemplo, neumonía por Pneumocystis jirovecii e infección por mycobacterium avium. Reacciones relacionadas con la infusión: Los eventos adversos asociados con la infusión (eventos seleccionados que se presentaron durante o dentro de las 24 horas de la infusión) fueron reportadas por el 4,5% de los pacientes en el grupo que recibió dosis de 8 mg/kg de ROACTEMRA® + FARME y en el 2,9% de los pacientes en el grupo tratados con placebo +FARME. Los eventos reportados durante la infusión fueron principalmente episodios de hipertensión; los eventos reportados dentro de las 24 horas de terminada una infusión fueron reacciones cutáneas (exantema, urticaria); dichos eventos no estuvieron limitados a la aplicación del tratamiento. Las reacciones anafilácticas (en 6 pacientes) u otras reacciones graves de hipersensibilidad asociadas con tocilizumab y que requirieron de la suspensión del tratamiento, fueron reportadas en un total de 12 de 3.728 pacientes (0,3%) tratados con ROACTEMRA®, durante los estudios clínicos abiertos y controlados. Esas reacciones se observaron durante la segunda a la quinta infusión de tocilizumab (ver Precauciones generales). Inmunogenicidad: un total de 1.747 pacientes han sido evaluados en busca de anticuerpos anti-tocilizumab en los estudios clínicos controlados. Veinticuatro pacientes (1,4%) desarrollaron anticuerpos anti-tocilizumab positivos, de lo cuales 4 (0,2%) tuvieron una reacción alérgica. En 18 pacientes (1%), quienes desarrollaron anticuerpos neutralizantes, no se observó una pérdida de eficacia hasta las 96 semanas de tratamiento.
Interacciones medicamentosas y de otro género: El análisis en la población para el estudio de farmacocinética reveló que el uso concomitante de los productos medicinales para la artritis reumatoide no influyó en la farmacocinética de ROACTEMRA®, tales como metotrexato, cloroquina y derivados, inmunosupresores (azatioprina, leflunomida), corticoesteroides (prednisona y derivados) ácido fólico y derivados, fármacos antiinflamatorios no esteroideos (diclofenaco, ibuprofeno, naproxen, meloxicam, inhibidores de la COX-2 (celecoxib), analgésicos (paracetamol, codeína, y derivados, tramadol). ROACTEMRA® no se ha estudiado en combinación con otros FARME biológicos.
La formación de las enzimas CYP3450 es suprimida por la inflamación crónica provocada por las citocinas. Por lo tanto se espera que para cualquier medicamento con un potente efecto antiinflamatorio, tal como ROACTEMRA®, la formación de las enzimas CYP450 podría normalizarse. Esto es clínicamente relevante para los sustratos CYP450 con un estrecho índice terapéutico, en donde la dosis se ajusta individualmente. Al iniciar la administración de ROACTEMRA®, en los pacientes que están siendo tratados con esos tipos de productos medicinales, deberá realizarse el monitoreo terapéutico del efecto (por ejemplo warfarina) o la concentración del fármaco (por ejemplo ciclosporina), y la dosis individual del producto medicinal deberá ajustarse según se requiera.
Alteraciones en los resultados de pruebas de laboratorio: Anormalidades en la biometría hemática: las disminuciones en la cuenta de neutrófilos por debajo de 1 x 109/l ocurrieron en el 3,4% de los pacientes tratados con 8 mg/kg de tocilizumab + FARME hasta < 0,1% de los pacientes tratados con placebo + FARME. Incremento en los niveles de las enzimas hepáticas: los incrementos transitorios en los niveles de ALT/ AST > 3 veces por arriba del límite normal, se observaron en el 2,1% de los pacientes tratados con 8 mg/kg de ROACTEMRA®, en comparación al 4,9% de los pacientes tratados con MTX, y en el 6,5% de los pacientes quienes recibieron 8 mg/kg de tocilizumab + FARME, en comparación al 1,5% de los pacientes que recibieron placebo + FARME. La adición de fármacos potencialmente hepatotóxicos (por ejemplo MTX) a la monoterapia con ROACTEMRA®, resultaron en un incremento en la frecuencia de esos incrementos. Los incrementos en los niveles de ALT/AST > 5 veces por arriba del límite normal se observaron en el 0,7% de los pacientes que estaban recibiendo la monoterapia con ROACTEMRA® y en el 1,4% de los pacientes que estaban recibiendo ROACTEMRA® + FARME, la mayoría de los cuales fueron retirados del tratamiento con ROACTEMRA®. Esos incrementos no se asociaron con ninguno de los incrementos clínicamente relevantes en los niveles de bilirrubina directa, ni tampoco se asociaron con la evidencia clínica de hepatitis o de insuficiencia hepática. Incrementos en los parámetros de lípidos: los incrementos en lípidos séricos (colesterol total, LDL, HDL, triglicéridos) se observaron en los pacientes tratados con ROACTEMRA®. En la mayoría de los pacientes, no hubo un incremento en el índices aterogénico y los incrementos en el colesterol total respondieron al tratamiento con agentes hipolipemiantes. Hipercolesterolemia: en los resultados de los estudios clínicos de ROACTEMRA®, se observó un incremento en el colesterol sérico en los grupos de pacientes que recibieron dicho medicamento. En el análisis de seguridad a largo plazo realizado a marzo 2008, se demostró que estas elevaciones se estabilizaron sin incrementos posteriores y únicamente 3,3% de los pacientes con elevación de colesterol inició terapia con agentes hipolipemiantes (p. ej. Estatinas), según las guías del ATP III (Adult Treatment Panel III) para el tratamiento de las hiperlipidemias. las cuales fueron efectivas para la reducción del colesterol total. Está bien establecido que los individuos con colesterol total elevado ( > 240 mg/dl) tienen un riesgo elevado de enfermedad cardiovascular y, por lo tanto, de eventos cardiovasculares. El Framingham Heart Study reportó que mujeres con niveles de colesterol sérico entre 200-239 mg/dl y ≥ 240 mg/dl tuvieron un riesgo relativo de desarrollar enfermedad de las arterias coronarias de 1,51 y 1,72, comparado con mujeres con colesterol total < 200 mg/dl (Wilson, 1998). Subsecuentemente, modificaciones en los hábitos dietéticos, ejercicio y terapias farmacológicas han demostrado ser efectivas en la reducción del colesterol total y reduciendo la incidencia de la enfermedad coronaria. Los pacientes tratados con ROACTEMRA® y que iniciaron terapia hipolipemiante sólo 10% había alcanzado el umbral de 130 mg/dl de colesterol-LDL que determina el inicio del tratamiento anti-hiperlipidemia. Dentro del análisis de seguridad, no se encontró evidencia que sugiera un incremento en el riesgo cardiovascular en los pacientes tratados con ROACTEMRA®. Es importante notar que la actividad de la enfermedad y los marcadores de la inflamación como la proteína C- reactiva (PCR) se correlacionan con un incremento en la incidencia de enfermedad coronaria. ROACTEMRA®es altamente efectivo en la reducción de la inflamación con una marcada reducción en la PCR. La incidencia de infarto del miocardio en los estudios clínicos es baja en los pacientes tratados con ROACTEMRA® comparada con el grupo control. Los pacientes que reciben tratamiento con estatinas para el control de la hipercolesterolemia pueden recibir tratamiento con ROACTEMRA® ya que en los estudios de farmacocinética y farmacodinamia demostraron que no hay interacciones medicamentosas entre estos fármacos y la eficacia de éstos se mantiene.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: no se ha realizado un estudio de carcinogenicidad de ROACTEMRA®. Los datos preclínicos disponibles mostraron la contribución de la citocina pleiotrópica IL-6 en la progresión maligna y la resistencia a la apoptosis de los diversos tipos de cáncer. Esos datos no sugieren un riesgo relevante para el inicio del cáncer y la progresión bajo la terapia con ROACTEMRA®. Consecuentemente, no se ha observado una proliferación de lesiones en un estudio de toxicidad crónica a 6 meses en el mono cynomolgus, ni tampoco se describieron en ratones con exceso IL-6 y bajo la depleción de IL-6 de manera crónica. Mutagenicidad: todos los estudios de genotoxicidad estándar con ROACTEMRA® en las células procarióticas y eucarióticas fueron negativos. Alteraciones en la fertilidad: no se realizaron estudios sobre fertilidad animal en especies relevantes. Sin embargo, los datos pre-clínicos no sugieren un efecto sobre la fertilidad bajo el tratamiento con ROACTEMRA®. Los efectos sobre los órganos endócrinos activos o sobre los órganos del sistema reproductor no se observaron en el estudio de toxicidad crónica en el mono cynomolgus, ni tampoco se afectó el desempeño reproductor en los ratones deficientes de IL-6. Teratogenicidad: cuando ROACTEMRA® se administró intravenosamente a los monos cynomolgus durante la gestación temprana, no se observaron efectos dañinos directos o indirectos sobre el embarazo o el desarrollo embrio-fetal. Otros: en un estudio de toxicidad embrio-fetal realizado en los monos cinomolgus, se observó un ligero incremento de aborto/fallecimiento embrio-fetal, con una elevada exposición sistémica ( > 100 veces la exposición humana) en el grupo tratado con dosis elevadas de 50 mg/kg/día, en comparación al grupo placebo y otros grupos con dosis bajas. Aun cuando la IL-6 no parece ser una citocina crítica para el crecimiento fetal o el control inmunológico de la interfase maternal /fetal, no se puede excluir una relación de este hallazgo con ROACTEMRA®.
Dosis y vía de administración: La dosis recomendada de ROACTEMRA® para los pacientes adultos es de 8 mg/kg, administrada una vez cada cuatro semanas como infusión IV. ROACTEMRA® se puede utilizar solo o en combinación con MTX y /u otros fármacos anti-reumáticos modificadores de la enfermedad (FARME). ROACTEMRA®deberá ser diluido hasta 100 ml, por un profesional al cuidado de la salud, con solución de cloruro de sodio estéril al 0,9%, utilizando una técnica aséptica (ver Recomendaciones para el almacenamiento). ROACTEMRA® se recomienda para Infusión IV durante 1 hora. Instrucciones para dosis especiales: niños: no se ha establecido la seguridad y eficacia de ROACTEMRA® en los niños. Ancianos: en los pacientes en edad avanzada, no se requiere de un ajuste en la dosis. Insuficiencia renal: no se requieren de ajustes en la dosis en los pacientes con insuficiencia renal (ver Farmacocinética en poblaciones especiales).Insuficiencia hepática: no se ha estudiado la seguridad y eficacia de ROACTEMRA® en los pacientes con insuficiencia hepática (ver Precauciones generales).
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Existen datos limitados disponibles acerca de la sobredosis con ROACTEMRA®. Se reportó un caso de sobredosis accidental, en donde el paciente con mieloma múltiple recibió una dosis única de 40 mg/kg. No se observaron reacciones adversas producidas por el fármaco. No se observaron reacciones adversas graves en los voluntarios sanos quienes recibieron una sola dosis de hasta 28 mg/kg, aun cuando se observó neutropenia limitada por la dosis.
Presentación(es): Caja con 1 frasco ámpula con 80mg/4 ml. Caja con 1 frasco ámpula con 200mg/10 ml. Caja con 1 frasco ámpula con 400mg/20 ml.
Recomendaciones sobre almacenamiento: Para los frascos ámpula: almacenar entre 2°C-8°C, no congelar. Para la solución de la infusión preparada: la solución de la infusión preparada de ROACTEMRA® es física y químicamente estable en solución de cloruro de de sodio al 0,9% a 30°C durante 24 horas. Desde el punto de vista microbiológico, la infusión preparada deberá utilizarse inmediatamente. De lo contrario, el almacenamiento y las condiciones antes de usarse son responsabilidad del usuario y no deberá ser mayor de 24 horas a 2-8°C, a menos que la dilución se haya realizado en condiciones asépticas controladas y validadas. Instrucciones especiales para su uso, manejo y desecho. Retirar la cantidad requerida de ROACTEMRA® (0,4 ml/kg) bajo condiciones asépticas y diluir hasta una concentración calculada de tocilizumab en una bolsa de infusión de 100 ml, conteniendo solución de cloruro de sodio al 0,9% estéril, no pirogénica. Para mezclar la solución, invertir suavemente la bolsa para evitar la formación de espuma. Los medicamentos administrados vía parenteral deben ser inspeccionados visualmente en busca de partículas extrañas o decoloración previa a la administración. Unicamente las soluciones claras a opalescentes, incoloras a amarillo pálido y libres de partículas visibles podrán ser infundidas.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni la lactancia. Consérvese en refrigeración, entre 2°C y 8°C. No se congele. Protéjase de la luz.
Nombre y domicilio del laboratorio: Hecho en Japón por: Chugai Pharma Manufacturing Co Ltd, 16-3 Kiyohara Kogyodanchi, Utsunomiya-city, Tochigi, Japón. Acondicionado y distribuido por: Productos Roche S.A. de C.V. Vía I. Fabela Nte. No. 1536-B, Col. Parque Industrial, 50030, Toluca, México. Para información adicional sobre este medicamento, comuníquese a nuestro centro de información médica, Tel. (01)(55) 52585099 y 01-800-8218887 o mexico.info@roche.com.
Número de registro del medicamento: 044M2009 SSA.
Clave de IPPA: BEAR-083300CT051574/R2009