ROACTEMRA SC
ROCHE
Denominación genérica: Tocilizumab.
Forma farmaceutica y formulación: Solución. Cada jeringa prellenada RoActemra® SC contiene: Tocilizumab 162 mg. Vehículo cbp 0.9 mL
Indicaciones terapéuticas: Artritis Regmatoide (AR)RoActemra SC está indicado para el tratamiento de la artritis reumatoide (AR) activa de intensidad moderada a grave, en pacientes adultos. RoActemra® SC se puede utilizar solo (en pacientes intolerantes al MTX o cuando no es apropiado continuar el tratamiento con MTX) o en combinación con metotrexato (MTX) y/u otros fármacos antirreumáticos modificadores de la enfermedad (FARME).
Farmacocinética y farmacodinamia: Farmacocinética: La farmacocinética de RoActemra® SC se determinó utilizando un análisis de población farmacocinética sobre una base de datos compuesta de 1,759 pacientes con artritis reumatoide tratados con 162 mg s.c. cada semana, 162 mg se. en semanas alternas y 8 mg/kg cada 4 semanas por 24 semanas. Los parámetros farmacocinéticos de RoActemra SC no cambiaron con el tiempo. Para la dosis de 162 mg cada semana, el equilibrio dinámico de la media pronosticada (±SD) AUC una semana, Cmin y Cmax de tocilizumab fue de 8,200 ± 3,600 mcg-h/ml, 44.6 ± 20.6 mcg/ml y 50.9 ± 21.8 mcg/ml, respectivamente. Los índices de acumulación para AUC, Cmin, y Cmax fueron de 6.83, 6.37, y 5.47, respectivamente. El equilibrio dinámico se alcanzó después de 12 semanas para AUC, Cmin, y Cmax. Para la dosis de 162mg en semanas alternas, el equilibrio dinámico de la media pronosticada (±SD) AUC2semanas, Cmin, y Cmax de tocilizumab fue de 3.200 ± 2,700 mcg-h/ml, 5.6 ± 7.0 mcg/ml y 12.3 ± 8.7 mcg/ml, respectivamente? Los índices de acumulación para AUC, Cmin, y Cmax fueron de 2.67, 5.6, y 2.12, respectivamente. El equilibrio dinámico se alcanzó después de 12 semanas para AUC y Cmin y después de 10 semanas para Cmax. Absorción: Después de la administración s.c. en los pacientes con artritis reumatoide, la semivida de absorción fue de aproximadamente 4 días. La biodisponibilidad para RoActemra® SC fue de 0.8. Distribución: Tocilizumab sufre eliminación bifásica desde la circulación. En los pacientes con artritis reumatoide el volumen central de distribución fue de 3.5 L, el volumen periférico de distribución fue de 2.9 L que da como resultado un volumen de distribución en equilibrio dinámico de 6.4L Eliminación: La depuración total de tocilizumab fue dependiente de la concentración y es la suma de la depuración lineal y la depuración no lineal. La depuración lineal se calculó como un parámetro en el análisis de población farmacocinética y fue de 12.5 mL/h en los pacientes con artritis reumatoide. La depuración no lineal dependiente de la concentración juega un papel importante a concentraciones bajas de tocilizumab. Una vez que la trayectoria de depuración no lineal se satura, a mayores concentraciones de tocilizumab, la depuración se determina principalmente por la depuración lineal. La t1/2 de tocilizumab es dependiente de la concentración en la artritis reumatoide. Para la administración intravenosa la t1/2 aparente dependiente de la concentración es de hasta 11 días para 4 mg/kg y de 13 días para 8 mg/kg cada 4 semanas en los pacientes con RA en fase estable.Para RoActemra® SC, Ia t1/2 aparente dependiente de la concentración es hasta de 13 días para 162 mg cada semana y de 5 días para 162 mg en semanas alternas en pacientes con AR en equilibrio dinámico. Farmacocinética en Poblaciones Especiales: Insuficiencia Hepática: No se ha realizado ningún estudio formal del efecto de la insuficiencia hepática sobre la farmacocinética de tocilizumab. Insuficiencia Renal: No se ha realizado ningún estudio formal del efecto de la insuficiencia renal sobre la farmacocinética de tocilizumab. La mayoría de los pacientes en la población con artritis reumatoide para el análisis de farmacocinética tuvieron una función renal normal o insuficiencia renal leve. La insuficiencia renal leve (depuración de creatinina basada en Cockcroft -Gault < 80 mL/min y ≥ 50 mL/min) no impactó la farmacocinética de tocilizumab. No se requiere ajuste de la dosis en pacientes con insuficiencia renal leve. Otras poblaciones esgeciales: El análisis farmacocinético de la población en pacientes adultos con artritis reumatoide mostró que la farmacocinética de tocilizumab no se ve afectada por la edad, el género o la raza. No es necesario ajustar la dosis para este tipo de factores demográficos. En los estudios clinicos con tocilizumab, se observó una rápida disminución en los niveles de proteína C reactiva (PCR), en la velocidad de sedimentación globular (VSG) y en la proteína amiloide A sérica. Se observó incremento en los niveles de hemoglobina, debido a que tocilizumab disminuye los efectos producidos por IL-6 sobre la producción de hepcidina y de esta manera incrementa la disponibilidad del hierro. En sujetos sanos a quienes se les administró tocilizumab en dosis de 2 a 28 mg/kg, la cuenta absoluta de neutrófilos disminuyó a su nivel mínimo en 3 a 5 días después de la administración. De ahí en adelante, hubo recuperación de neutrófilos hacia el límite basal, de forma dependiente de la dosis. Los pacientes con artritis reumatoide, mostraron un patrón similar de cuenta absoluta de neutrófilos después de la administración de tocilizumab (v. Alteraciones de pruebas de laboratorio). Mecanismo de Acción: RoActemra® SC es un anticuerpo monoclonal dirigido contra el receptor de la interleucina-6 (IL-6) antihumana, humanizada recombinante de la subclase de inmunoglobulina (lg) IgG1. RoActemra® SC se une específicamente a los receptores de IL-6 tanto solubles como los unidos a la membrana (sIL-6R y mIL-6R), y ha demostrado que inhibe la cascada de señalización mediada por sIL-6R y mIL-6R. La IL-6 es una citocina multifuncional, producida por una gran variedad de tipos de células involucradas en la función paracrina local así como la regulación de los procesos fisiológicos y patológicos sistémicos tales como la inducción de la secreción de inmunoglobulinas, la activación de linfocitos T, la inducción de proteínas hepáticas de fase aguda y la estimulación de hematopoyesis. La IL-6 ha sido implicada en la patogénesis de varias enfermedades, incluyendo las enfermedades inflamatorias, la osteoporosis y neoplasias. Existe la posibilidad de que tocilizumab afecten a las defensas del huésped frente a infecciones y neoplasias malignas. El papel de la inhibición del receptor IL-6 en el desarrollo de cáncer no se conoce. Estudios de Eficacia Clínica: Se evaluó la eficacia de RoActemra® SC administrado subcutáneamente en un estudio multicéntrico doble ciego controlado en pacientes con AR activa. El estudio (SC-I) requirió que los pacientes fueran > 18 años de edad con artritis reumatoide activa diagnosticada de acuerdo con los criterios de la ACR y tuvieran por lo menos 4 articulaciones dolorosas a la palpación y 4 articulaciones inflamadas al inicio del estudio. Todos los pacientes recibieron FARMEs no biológicos de fondo. El estudio SC-l evaluó pacientes con artritis reumatoide activa de intensidad moderada a severa que tuvieron una respuesta clínica inadecuada a su tratamiento reumatológico existente, incluyendo uno o más FARMEs. Aproximadamente el 20% tuvo un antecedente de respuesta inadecuada a por lo menos un inhibidor del Factor de necrosis tumoral (aFNT. En el estudio SC-I, 1,262 pacientes fueron aleatorizados en una proporción 1:1 para recibir 162 mg de RoActemra® SC cada semana u 8 mg/kg de tocilizumab ¡.v. cada cuatro semanas combinado con FARME no biológicos. El criterio principal de valoración en el estudio fue la diferencia en la proporción de pacientes que alcanzaron una respuesta ACR20 a la semana 24. Los resultados del estudio SC-I se muestran en la Tabla 1.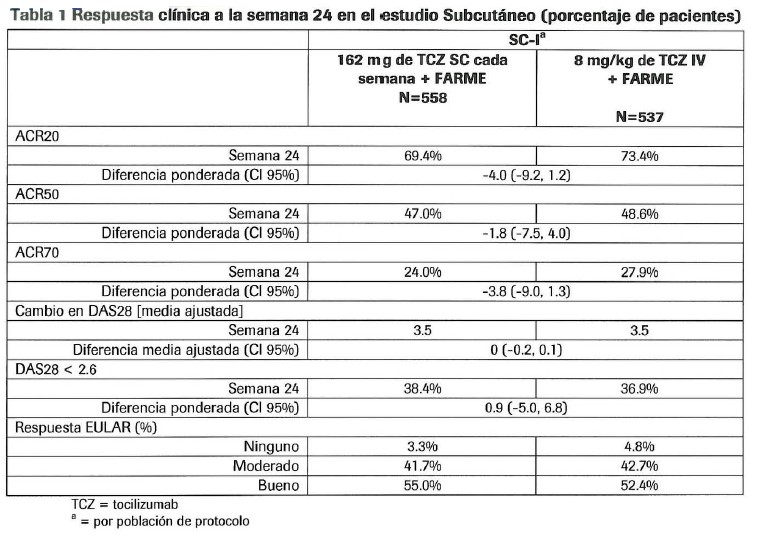
Respuesta radiográfica: La respuesta radiográfica de tocilizumab administrado subcutáneamente se evaluó en un estudio multicéntrico controlado doble ciego en pacientes con AR activa. Este estudio (SC-ll) evaluó pacientes con artritis reumatoide activa de intensidad moderada a severa que tuvieron una respuesta clínica inadecuada a su tratamiento reumatológico existente, incluyendo uno o más FARMES donde aproximadamente el 20% tuvo un antecedente de respuesta inadecuada a por lo menos un inhibidor del FNT. Se requirió que los pacientes fueran > 18 años de edad con artritis reumatoide activa diagnosticada de acuerdo con los criterios de la ACR y que por lo menos tuviera 8 articulaciones dolorosas a la palpación y 6 articulaciones inflamadas al inicio del estudio. En SC-II, fueron aleatorizados 656 pacientes en una proporción 2:1 a 162 mg de RoActemra® SC en semanas alternas o a placebo en combinación con FARME no biológicos. En el estudio SC-Il, se evaluó radiológicamente la inhibición del daño articular estructural y se expresó como un cambio de la inicial en la puntuación Sharp total media modificada de van der Heijde (mTSS). En la semana 24, se mostró la inhibición del daño estructural, con evolución radiográfica considerablemente menor en pacientes que recibieron tocilizumab s.c. comparado con placebo (mTSS de 0.62 vs. 1.23, p=0.0149 (van Elteren). Estos resultados concuerdan con los obervados en pacientes tratados con tocilizumab intravenoso. El estudio SC-II fue un estudio clínico puente a los estudios clínicos de la vía de administración IV, tanto para seguridad como para eficacia, por tanto los resultados que a continuación se muestran para la vía de administración IV son válidos también para la vía subcutánea. Resultados de la Calidad de Vida: En el estudio SC-I, la disminución media en HAQ-DI desde la inicial hasta la semana 24 fue del 0.6 tanto para 162 mg de tocilizumab s.c. semanalmente como para 8 mg/kg de tocilizumab i.v. cada 4 semanas. La proporción de pacientes que lograron una mejora clínicamente relevante en HAQ-DI a la semana 24 (cambio desde la inicial de ≥ 0.3 unidades) fue comparable en el grupo de tocilizumab s.c. cada semana (65.2%) comparado con el grupo de 8 mg/kg de tocilizumab i.v. (67.4%), con una diferencia ponderada en las proporciones de -2.3% (Cl 95% -8.1, 3.4). El resumen de SF-36 se dividió en componentes mentales y físicos. Las puntuaciones de componente mental fueron similares entre los grupos con un cambio medio desde la inicial a la semana 24 de 6.22 para el grupo se y de 6.54 para el grupo i.v. Las puntuaciones de componente físico también fueron similares entre los grupos, con un cambio medio desde la inicial a la semana 24 de 9.49 para el grupo 3.0. y de 9.65 para el grupo i.v.
Contraindicaciones: Hipersensibilidad a Tocilizumab o a cualquiera de los excipientes. Tocilizumab está contraindicado en menores de 18 años. Durante el embarazo y perdida de lactancia.
Precauciones generales: General: Para mejorar la rastreabilidad de medicamentos biológicos, el nombre comercial del producto administrado debe registrarse (o mencionarse) claramente en el expediente del paciente. Infecciones: Se han reportado infecciones graves y algunas veces mortales en pacientes que están recibiendo agentes inmunosupresores incluyendo tocilizumab (v. Reacciones secundarias y adversas). El tratamiento con RoActemra® SC no se debe iniciar en los pacientes con infecciones activas graves. La administración de RoActemra® SC deberá interrumpirse si un paciente desarrolla una infección grave, hasta que la infección sea controlada. Los médicos deben tener precaución cuando consideren utilizar RoActemra® SC en los pacientes con historial de infección recurrente o condiciones subyacentes (por ejemplo diverticulitis, diabetes), lo cual puede predisponer a los pacientes a infecciones. Se recomienda la vigilancia para la detección oportuna de una infección grave en los pacientes que están recibiendo tratamientos biológicos para la artritis reumatoide de intensidad moderada a severa, debido a que los signos y síntomas de la inflamación aguda pueden disminuirse, asociados con la supresión de reactantes de fase aguda. Complicaciones de diverticulitis: Se han reportado eventos de perforación gastrointestinal como complicacion de la diverticulitis en pacientes con AR. RoActemra® SC debe utilizarse con precaución en pacientes con historia previa de ulceración intestinal o diverticulitis. Los pacientes que presentan síntomas potencialmente indicativos de diverticulitis complicada, tales como dolor abdominal, deben ser evaluados rápidamente para la identificación temprana de diverticulitis y subsecuente perforación gastrointestinal. Tuberculosis: Como se recomienda para otras terapias biológicas para artritis reumatoide, los pacientes deben ser examinados para detección de tuberculosis latente antes de iniciar el tratamiento con RoActemra® SC. Los pacientes con tuberculosis latente deben ser tratados con la terapia estándar antimicobacteriana antes de iniciar tratamiento con RoActemra® SC. Es necesario valorar el perfil riesgo-beneficio para el paciente y mantener un monitoreo constante así como informarle de los signos y síntomas de Tuberculosis. Vacunas Las vacunas vivas y atenuadas no se deberán administrar concurrentemente con RoActemra® SC, debido a que no se ha establecido la seguridad clínica. No existen datos disponibles sobre la transmisión secundaria de la infección por las personas que están recibiendo vacunas vivas, a los pacientes que están recibiendo tocilizumab. Se recomienda que todos los pacientes, se actualicen con todas las inmunizaciones de acuerdo a las guías de inmunización vigentes antes de iniciar la terapia con RoActemra® SC. El intervalo entre las vacunas vivas y el inicio de la terapia con RoActemra® SC debe estar en concordancia con las guías vigentes de vacunación con respecto a los agentes inmunosupresores. Reacciones de Hipersensibilídad: Reacciones de hipersensibilidad graves, incluyendo anafilaxia, han sido reportadas en asociación con la administración de tocilizumab (v. Reacciones Adversas). En el marco de post-comercialización eventos de hipersensibilidad grave y anafilaxia han ocurrido en pacientes tratados con un rango de dosis de Tocilizumab con o sin terapias concomitantes para artritis, la premedicación, y/o una reacción previa de hipersensibilidad. Si se produce una reacción anafiláctica u otra reacción de hipersensibilidad grave, la administración de RoActemra® SC se debe detener de inmediato y RoActemra® SC debe suspenderse de forma permanente. Enfermedad Hepática Activa e Insuficiencia Hepática: El tratamiento con tocilizumab, particularmente cuando se administra concomitantemente con metotrexato, se puede asociar con incrementos en los niveles de transaminasas hepáticas, por lo tanto se debe tener precaución al considerar el tratamiento de los pacientes con enfermedad hepática activa o insuficiencia hepática (v. Instrucciones para dosis especiales y Alteraciones de pruebas de Laboratorio). Reactividad viral: Se ha reportado reactivación viral (ejem: virus Hepatitis B) con terapias biológicas para el tratamiento de artritis reumatoide. En estudios clínicos con tocilizumab, se excluyeron a los pacientes positivos a hepatitis. Es necesario realizar un monitoreo estrecho a fin de detectar cualquier dato de infección activa. Trastornos desmielinizantes: Los médicos deben estar alertas de síntomas potencialmente indicativos de trastornos centrales demielinizantes de reciente inicio. Aún se desconoce el potencial de RoActemra® SC en relación con desórdenes cognitivos centrales. Es necesario evaluar el perfil riesgo-beneficio en pacientes con antecedentes, riesgo elevado o patologías desmielinizantes. Abuso y Dependencia de Fármacos: No se han realizado estudios sobre los efectos en el potencial de RoActemra® SC para producir dependencia. Sin embargo, no existe evidencia de los datos disponibles de que el tratamiento de RoActemra® resulta en dependencia. Capacidad para Conducir y Uso de Maquinaria: No se han realizado estudios sobre la capacidad para conducir y el uso de maquinaria. Sin embargo, no existe evidencia a partir de los datos disponibles, de que el tratamiento con RoActemra® SC afecta la capacidad para conducir y el uso de máquinas. Pruebas de laboratorio: Neutropenia: El tratamiento con tocilizumab se asoció con una mayor incidencia de neutropenia. En estudios clínicos no se asoció la neutropenia relacionada con el tratamiento con infección grave (Alteraciones de pruebas de laboratorio). Debe tenerse precaución cuando se considere el inicio del tratamiento con RoActemra® SC en pacientes con un número bajo de neutrófilos, es decir, número absoluto de neutrófilos (ANC) < 2 x 109/L. En pacientes con un número absoluto de neutrófilos < 0.5 x 109/I, no se recomienda el tratamiento. En AR, deben monitorearse los neutrófilos de 4 a 8 semanas después de iniciar el tratamiento, de acuerdo con las buenas prácticas clínicas. Para las modificaciones recomendadas a la dosis basada en los resultados de ANC, consulte la sección dosis y vía de administración. Trombocitapenia: El tratamiento con tocilizumab se asoció con una reducción en el número de plaquetas. La reducción relacionada con el tratamiento en las plaquetas no se asoció con eventos de sangrado grave en estudios clínicos (v. Alteraciones de pruebas de laboratorio). Debe tenerse precaución cuando se considere iniciar el tratamiento de RoActemra® SC en pacientes con un número de plaquetas por debajo de 100 x 10 3/mL. En pacientes con número de plaquetas < 50 x 103/ mL, no se recomienda el tratamiento. En AR, deben monitorearse las plaquetas de 4 a 8 semanas después de iniciar el tratamiento, de acuerdo con las buenas prácticas clínicas. Para las modificaciones recomendadas a la dosis basada en el número de plaquetas, consulte la sección dosis y vía administración. Elevaciones en las transaminasas hepáticas: En estudios clínicos, se observaron elevaciones de leves a moderadas en las transaminasas hepáticas con el tratamiento tocilizumab, sin evolución a lesión hepática (v. Alteraciones de pruebas de laboratorio). Se observó incremento en la frecuencia de estas elevaciones cuando se utilizaron potenciales medicamentos hepatotóxicos (por ejemplo, metotrexato [MTX]] en combinación con tocilizumab. Debe tenerse precaución cuando se considere iniciar el tratamiento de RoActemra® SC en pacientes con transaminasas ALT o AST > 1.5x ULN (por encima del límite superior normal, por sus siglas en inglés) elevadas. En pacientes con ALT o AST > 5x ULN elevadas, no se recomienda el tratamiento. En AR, debe monitorearse ALT y AST de 4 a 8 semanas después de iniciar el tratamiento de acuerdo con las buenas prácticas clínicas. Para las modificaciones recomendadas a la dosis basada en transaminasas, v. Dosis y vía administración. Parámetros de lípidos: Se han observado elevaciones en los parámetros de lípidos como son colesterol, triglicéridos y/o colesterol de lipoproteína de baja densidad (LDL) (v. Alteraciones de pruebas de laboratorio). En AR, debe realizarse la evaluación de parámetros de lípidos de 4 a 8 semanas después de iniciar el tratamiento con RoActemra® SC. Deben manejarse a los pacientes de acuerdo con las directrices clínicas locales para el manejo de hiperlipidemia.
Restricciones de uso durante el embarazo y lactancia: Embarazo: No existen datos adecuados del uso de tocilizumab en las mujeres embarazadas. Un estudio realizado en monos no indicó ningun potencial dismorfogenético, pero alcanzó un alto número de abortos espontáneos/fallecimientos embrio-fetales con la dosis elevada. Se desconoce la relevancia de estos datos para los humanos. Tocilizumab no deberá utilizarse durante el embarazo. Lactancia: No se sabe si tocilizumab es excretado en la leche humana. Aun cuando las inmunoglobulinas endógenas del isotipo IgG son secretadas en la leche humana, es improbable una absorción sistémica de tocilizumab por medio del amamantamiento debido a una rápida degradación proteolítica de dichas proteínas en el sistema digestivo? Tocilizumab no deberá administrarse durante el periodo de lactancia.
Reacciones secundarias y adversas: Estudios Clínicos: Pacientes tratados con tocilizumab subcutáneo: La seguridad de RoActemra® SC (subcutáneo) en AR se estudió en SC-I. El estudio comparó la eficacia y seguridad de 162 mg de RoActemra® SC administrado cada semana s.c. comparado con RoActemra® (intravenoso) a 8 mg/kg i.v. en 1,262 pacientes con AR adulta. Todos los pacientes en el estudio recibieron FARMEs no biológicos de fondo. La seguridad e inmunogenicidad observada para RoActemra® SC administrado subcutaneamente concordó con el perfil de seguridad conocido de tocilizumab ¡.V. y no se observaron reacciones adversas al medicamento nuevas o inesperadas. Se observó una mayor frecuencia de reacciones en el sitio de inyección en los grupos s.c. comparado con las inyecciones se de placebo en los grupos i.v. (v. eficacia clínica). Reacciones en el sitio de inyección: Durante el período controlado de 6 meses, en SC-l, la frecuencia de las reacciones en el sitio de inyección fueron del 10.1% (64/631) y del 24% (15/631) para inyecciones semanalmente de RoActemra® SC y placebo s.c. (grupo i.v.), respectivamente. Estas reacciones en el sitio de inyección (incluyendo eritema, prurito, dolor y hematoma) fueron de leves a moderadas en intensidad. La mayoría se resolvió sin ningún tratamiento y no requirieron suspensión del medicamento. Inmunogenícidad: En SC-l, un total de 625 pacientes tratados con 162 mg de RoActemra® SC semanalmente se sometieron a prueba para anticuerpos anti tocilizumab en el período controlado de 6 meses. Cinco pacientes (0.8%) presentaron anticuerpos anti tocilizumab, todos presentaron anticuerpos anti tocilizumab neutralizantes. Un total de 1,454 de todos los pacientes expuestos a RoActemra® SC se han sometido a prueba para anticuerpos anti tocilizumab, trece pacientes (0.9%) fueron positivos para anticuerpos anti tocilizumab y de estos 12 pacientes (0.8%) presentaron anticuerpos anti tocilizumab neutralizantes. Se observó correlación del anticuerpo, desarrollo a la respuesta clínica o evento adverso. Los estudios SC-I y SCII fueron estudios clínicos puente a los estudios clínicos de la vía de administración IV. tanto para seguridad como para eficacia, por tanto los resultados que a continuación se muestran para la vía de administración IV son válidos también para la vía subcutánea.
Interacciones medicamentosas y de otro género: En los análisis farmacocinéticos poblacionales no se detectó ningún efecto del MTX, medicamentos antiinflamatorios no esteroideos o corticosteroides sobre la depuración de tocilizumab. La administración concomitante de una dosis única de 10 mg/kg tocilizumab con 10-25 mg de MTX una vez a la semana no tuvo ningún efecto clínicamente significativo sobre la exposición MTX. Tocilizumab no se ha estudiado en combinación con otros FARMEs biológicos. La expresión de las enzimas CYP450 es suprimida por la inflamación crónica provocada por las citocinas. Por lo tanto se espera que para cualquier medicamento con un potente efecto anti-inflamatorio, tal como tocilizumab, la formación de las enzimas CYP450 podría normalizarse. En estudios in vitro con hepatocitos humanos cultivados demostrando que la IL-6 causó una reducción de la CYP1A2, CYP2C9, CYP2C19, y la expresión de la enzima CYP3A4. Tocilizumab normaliza la expresión de estas enzimas. El efecto de tocilizumab en enzimas CYP (excepto CYP2C19 y CYP2D6) es clínicamente relevante para los sustratos CYP450 con un índice terapéutico estrecho, y/o donde la dosis se ajusta individualmente. En un estudio en pacientes con AR, los niveles de simvastatina (CYP3A4) disminuyeron en un 57 % unas semanas después de una dosis única de tocilizumab, a un nivel similar o ligeramente superior que las observadas en sujetos sanos. Al iniciar o interrumpir el tratamiento con tocilizumab, los pacientes que toman medicamentos, que son individualmente ajustados a la dosis y metabolizan a través del CYP45O 3A4, 1A2 ó 2C9 (por ejemplo, atorvastatina, bloqueadores de los canales de calcio, teofilina, warfarina, fenitoína, ciclosporina, o benzodiazepinas) debe ser monitoreada ya que las dosis de estos productos puede necesitar ser ajustada para mantener su efecto terapéutico. Debido a su larga vida media de eliminación (tl/2), el efecto de tocilizumab sobre la actividad de las enzimas CYP450 puede persistir durante varias semanas después de interrumpir la terapia.
Alteraciones en los resultados de pruebas de laboratorio: Anormalidades en la biometría hemática: Neutrófilos: Administración Subcutánea: Durante el monitoreo de rutina de laboratorio de los estudios clínicos controlados a 6 meses de SC-I de RoActemra SC, se presentó una disminución en el número de neutrófilos por debajo de 1 x 109/L en el 2.9% de pacientes con 162 mg de RoActemra® SC semanalmente. No hubo una clara relación entre las disminuciones de neutrófilos por debajo de 1 x 109/L y la incidencia de infecciones graves. Plaquetas: Administración Subcutánea: Durante el monitoreo de rutina de laboratorio de los estudios clínicos controlados a 6 meses de SC-I de RoActemra® SC, ninguno de los pacientes tuvo una disminución en el número de plaquetas ≤550 x 10 3/mL. Incremento en los niveles de las enzimas hepáticas: Administración Subcutánea: Durante el monitoreo de rutina de laboratorio de los estudios clínicos controlados a 6 meses de SC-l de RoActemra® SC, se presentó la elevación en ALT o AST ≤3 x ULN en el 6.5% y en el 1.4% de pacientes, respectivamente en s.c. semanalmente. Incrementos en los parámetros de lípidos: Administración Subcutánea: Durante el monitoreo de rutina de laboratorio de los estudios clínicos controlados a 6 meses de SC-I de RoActemra® SC, el 19% de los pacientes con s.c. semanalmente experimentaron elevaciones sostenidas en el colesterol total > 6.2 mmoI/I (240 mg/dl) y el 9% presentó un incremento sostenido de LDL ≥ 4.1 mmoI/I (160 mg/dl) en s.c. semanalmente.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: No se ha realizado un estudio de carcinogenicidad de RoActemra®. Los datos pre-clínicos disponibles mostraron la contribución de la citocina pleiotrópica lL-6 en la progresión maligna y la resistencia a la apoptosis de los diversos tipos de cáncer. Esos datos no sugieren un riesgo relevante para el inicio del cáncer y la progresión bajo la terapia con RoActemra®. Consecuentemente, no se ha observado una proliferación de lesiones en un estudio de toxicidad crónica a 6 meses en el mono cynomolgus, ni se describieron en ratones con exceso lL-6 y bajo la depleción de IL-6 de manera crónica. Mutagenícidad: Todos los estudios de genotoxicidad estándar con RoActemra® en las células procarióticas y eucarióticas fueron negativos. Alteraciones en la fertilidad: Los datos pre-clínicos no sugieren un efecto sobre la fertilidad bajo el tratamiento con un análogo de RoActemra®. Los efectos sobre los órganos endocrinos activos o sobre los órganos del sistema reproductor no se observaron en el estudio de toxicidad crónica en el mono cynomolgus, ni tampoco se afectó el desempeño reproductor en los ratones machos y hembras deficientes de IL-6. Teratogenicidad: Cuando se administró RoActemra® intravenosamente a los monos cynomolgus durante la gestación temprana, no se observaron efectos dañinos directos o indirectos sobre el embarazo o el desarrollo embrio-fetal. Otros: En un estudio de toxicidad embrio-fetal realizado en los monos cynomolgus, se observó un ligero incremento de aborto/fallecimiento embrio-fetal, con una elevada exposición sistémica ( > 100 veces la exposición humana) en el grupo tratado con dosis elevadas de 50 mg/kg/día, en comparación al grupo placebo y otros grupos con dosis bajas. La incidencia de aborto fue en los antecedentes históricos para el mono cynomo/gus en cautiverio y los casos individuales de muerte aborto aborto/embrio fetal no mostraron ninguna relación consistente para la dosis o la duración en la dosis con RoActemra®. Aun cuando la lL-6 no parece ser una citocina crítica para el crecimiento fetal o el control inmunológico de la interfase maternal /fetal, no se puede excluir una relación de este hallazgo con RoActemra®. Se ha observado transferencia del análogo murino de RoActemra® a la leche en ratas en periodo de lactancia. El tratamiento con un análogo murino no ejerció toxicidad en ratones jóvenes. En particular, no hubo afectación del crecimiento óseo, función inmunologica y maduración sexual. El perfil de seguridad no clínica de tocilizumab en el macaco no sugiere una diferencia entre las vías de administración s.c. (RoActemra® SC) e i.v. (RoActemra®).
Dosis y vía de administración: General: La sustitución por otros medicamentos biológicos requiere el consentimiento del médico tratante. Para pacientes adultos con AR, RoActemra® SC debe administrarse como una inyección subcutánea. La formulación de RoActemra® SC se administra como una jeringa prellenada + dispositivo seguro de aguja (PFS+NSD) para un solo uso. La primera inyección debe realizarse bajo la supervisión de un profesional de la salud. Los sitios de inyección recomendados (abdomen, muslo y parte superior del brazo) deben rotarse y las inyecciones nunca deben administrarse en lunares, cicatrices o áreas donde la piel sea dolorosa a la palpación, amoratada, roja, dura o no intacta. Régimen de administración subcutánea: La dosis recomendada de RoActemra® SC para pacientes adultos es de 162 mg administrada una vez a la semana como una inyección subcutánea. RoActemra® SC puede usarse solo o en combinación con MTX y/o otros FARMEs. Los pacientes que cambian del tratamiento de tocilizumab i.v. (RoActemra) a la administración subcutánea (RoActemra® SC) deben administrar la primera dosis subcutánea al momento de la siguiente dosis intravenosa programada bajo la supervisión de un profesional de la salud calificado. La formulación de RoActemra® SC no está indicada para administración intravenosa. Evalúe la idoneidad del paciente para usar RoActemra® SC en casa y solicite a los pacientes de informar al profesional de la salud si presentan síntomas de reacción alérgica antes de administrar las siguientes dosis. Los pacientes deben buscar atención médica inmediata si presentan síntomas de reacciones alérgicas graves (v. Precauciones Generales). Recomendaciones para la modificación de la dosis para AR: (v. Pruebas de laboratorio).


Instrucciones para dosis especiales: Niños: La formulación de RoActemra SC no está recomendada para uso en niños menores de 18 años de edad debido a la escasez de datos sobre seguridad y eficacia. Ancianos: En los pacientes en edad avanzada no se requiere de un ajuste en la dosis. Insuficiencia Renal: No se requieren de ajustes en la dosis, en los pacientes con insuficiencia renal (v. Farmacocinética en poblaciones especiales). RoActemra°'D SC no se ha estudiado en pacientes con daño renal de moderado a severo. Insuficiencia Hegática: No se ha estudiado la seguridad y eficacia de RoActemra® SC en los pacientes con insuficiencia hepática (v. Precauciones generales).
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Existen datos limitados disponibles acerca de la sobredosis con tocilizumab. Se reportó un caso de sobredosis accidental, en donde el paciente con mieloma múltiple recibió una dosis única de 40 mg/kg i.v. No se observaron reacciones adversas producidas por el fármaco. No se observaron reacciones adversas graves en los voluntarios sanos quienes recibieron una sola dosis de hasta 28 mg/kg i.v., aun cuando se observó neutropenia limitada por la dosis.
Presentaciones: Caja con 4 jeringas prellenadas con 162mg/0.9mL
Recomendaciones para el almacenamiento: El medicamento no debe usarse después de la fecha de caducidad mostrada en la jeringa prellenada y el empaque. Almacene la jeringa prellenada en un refrigerador a una temperatura de 2 a 8°C (36 a 46°F). No congele, mantenga en la caja para protegerlo de la luz y mantenerlo seco. Instrucciones especiales para su uso, manejo y desecho. No use si el medicamento está turbio o contiene partículas, si existe algún color fuera de incoloro a color amarillento, o cualquier parte de la jeringa prellenada parece estar dañada. Desecho de las jeringas/objetos punzantes. Se debe cumplir estrictamente con los siguientes puntos en cuanto al uso desecho de la de la jeringa prellenada: Nunca se deben volver a utilizar las jeringas. Coloque todas las jeringas en un recipiente para objetos punzantes [recipiente desechable a prueba de punción). Mantenga este recipiente fuera del alcance de los niños. Debe evitarse colocar los recipientes para objetos punzantes usados en el basurero de la casa. Deseche el Recipiente completo oe acuerdo con los requerimientos locales o como lo instruya su proveedor sanitario. Para uso en casa, los pacientes deben procurar tener un recipiente resistente a la punción para desechar las jeringas usadas.
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni la lactancia. Consérvese en refrigeración, entre 2 °C y 8 °C. No se congele. Protéjase de la luz. Reporte las sospechas de reacción adversa al correo: farmacovigliancia@cofepris.gob.mx. También reporte las sospechas de reacciones adversas a través del centro de información médica y la unidad de farmacovigilancia de Roche: Tels: [55] 52585225. 52585099, 01800821887. Fax: [55] 52585475. Correo: mexico.info@roche.com.
Nombre del laboratorio y dirección: Importado, acondicionado y distribuido por: Productos Roche, S.A. de CV. Vía Isidro Fabela Nte. No. 1536-B, Col. Parque Industrial. CP. 50030, Toluca, México.
Número de registro del medicamento: 302M2015.