SELZENTRY
GSK
Denominación genérica: Maraviroc.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: Maraviroc 150 mg y 300mg. Excipiente cbp 1 Tableta.
Indicaciones terapéuticas: SELZENTRY® en combinación con otros medicamentos antirretrovirales, está indicado para los pacientes adultos previamente tratados, infectados sólo por el VIH-1 CCR-5 trópico. (Ver Dosis y Vía de Administración). Esta indicación está basada en los resultados de seguridad y eficacia de dos ensayos clínicos doble ciego, controlados con placebo, en pacientes que previamente ya habían recibido tratamiento (Ver Estudios Clínicos).
Farmacocinética y farmacodinamia: Farmacodinamia: Código ATC: Grupo farmacoterapéutico: Antivirales para uso sistémico, otros antivirales código ATC code J05AX09. Mecanismo de acción: El maraviroc pertenece a una clase terapéutica llamada antagonistas del co- receptor CCR5. Maraviroc se une selectivamente al receptor de la quimioquina CCR5, impidiendo la entrada del VIH-1 CCR5 a las células. Efectos farmacodinámicos: Actividad antiviral en cultivos celulares: El valor EC90 ajustado en suero, en las 43 cepas clínicas primarias aisladas de VIH-1 con tropismo fue 0.57 (0.06 - 10.7) ng/ML (fracción no unida), sin cambios significativos entre los diferentes subtipos evaluados. Maraviroc no muestra actividad antiviral en cultivos celulares en contra de los virus que pueden utilizar al CXCR4 como su co-receptor de entrada (virus con tropismo dual o tropismo a CXCR4), llamados colectivamente en adelante como "Virus usuarios del CXCR4". La actividad antiviral del contra el VIH-2 no se ha evaluado. Cuando se utilizó con otros medicamentos antirretrovirales en cultivos celulares, la combinación del maraviroc no fue antagónica con diferentes ITRNs (inhibidores de la transcriptasa reversa nucleósidos), ITRNNs (inhibidores de la transcriptasa reversa no nucleósidos) IPs (inhibidores de proteasa) o el inhibidor de fusión del VIH, enfuvirtida. Resistencia: Puede ocurrir escape viral de maraviroc por 2 vías: la selección de virus, que pueden utilizar al CXCR4 como su co-receptor de entrada (virus usarios del CXCR4) o la selección de virus que continúan utilizando exclusivamente el CCR5 (virus usarios del CCR5). Resistencia en cultivos celulares: Las variantes del VIH-1 con sensibilidad reducida al maraviroc se han seleccionado en cultivos celulares, posterior al paso serial de dos aislamientos clínicos de cepas VIH-1 con tropismo a CCR5. Los virus resistentes al maraviroc permanecieron con tropismo a CCR5 y no se observó una conversión de los virus trópicos a CCR5 o a virus con tropismo al receptor CXCR4. Resistencia fenotípica: las curvas de respuesta-concentración, para los virus resistentes al maraviroc, se caracterizaron como curvas que no alcanzaron el 100% de inhibición, en estudios que emplearon diluciones seriadas de maraviroc. El resultado del número de cambios de la EC50 tradicional no fue un parámetro útil para la medición de la resistencia fenotípica, ya que esos valores algunas veces no presentaron cambios a pesar de la sensibilidad significativamente reducida. Resistencia genotípica: se encontró que las mutaciones se acumulaban en la capa de la glicoproteína gp120 (la proteína viral que se une al co-receptor CCR5). La posición de estas mutaciones no fue consistente entre las diferentes cepas aisladas. De ahí que se desconoce la relevancia de estas mutaciones para la susceptibilidad a maraviroc en otros virus. Resistencia cruzada: las cepas clínicas aisladas de VIH-1, resistentes a ITRN, a ITRNN, IP y al enfuvirtida fueron susceptibles a maraviroc en cultivos celulares. Los virus resistentes al maraviroc, que se presentaron en cultivos celulares, permanecieron sensibles al inhibidor de fusión enfuvirtida y al inhibidor de proteasa saquinavir. In vivo: Ambas vías de resistencia se observaron en los estudios clínicos en pacientes con tratamiento previo. En la falla virológica, la presencia de virus con tropismo CXCR4, parece originarse de una población viral preexistente. Realizar pruebas de tropismo antes de la terapia para detectar la presencia de esta forma viral, puede reducir la incidencia de fracaso a través de este mecanismo. En pacientes donde falló la terapia antirretroviral, infectados sólo con virus R5, el maraviroc podría considerarse todavía activo si el valor de inhibición porcentual máxima (IPM) es de ≥95% (ensayo de Phenosense Entry). No se ha determinado la actividad residual in vivo de virus con valores IPM < 95%. La resistencia del virus R5 a través del aumento del EC50 no parece ser un mecanismo importante de fracaso terapéutico. Resistencia genotípica: en la actualidad, no es posible sugerir mutaciones clave (región V3) debido a la alta variabilidad de la secuencia V3 y al bajo número de muestras analizadas. Pacientes con tratamiento previo: En los estudios (MOTIVATE 1 y MOTIVATE 2), en 7.6% de los pacientes hubo un cambio en el resultado del tropismo: de tropismo a CCR5 a tropismo CXCR4 o a tropismo dual/mixto entre la exploración y la línea basal. Fracaso con virus con tropismo al receptor CXCR4: En aproximadamente el 55% de los sujetos en los que falló el tratamiento con maraviroc se detectaron virus con tropismo al receptor CXCR4, en comparación con el 6% de los sujetos que experimentaron fracaso en el tratamiento en el grupo de terapia de base optimizada (TBO). Con el fin de investigar el origen probable de los virus con tropismo a CXCR4 en el tratamiento, se realizó un análisis clonal detallado del virus en una muestra representativa de 20 sujetos (16 sujetos de los grupos de maraviroc y 4 sujetos del grupo con TBO solo) en los que se detectaron virus con tropismo CXCR4. Este análisis indicó que los virus con tropismo CXCR4 surgieron de un reservorio pre-existente no detectado en el análisis basal, en vez de que provinieran de la mutación de los virus con tropismo a CCR5 presentes en la basal. El análisis de tropismo, después de la falla terapéutica con maraviroc por virus con tropismo CXCR4 en pacientes con resultado basal de virus con tropismo a CCR5, demostró que la población viral revirtió al tropismo a CCR5 en 33 de los 36 pacientes con más de 35 días de seguimiento. Al momento del fracaso con los virus con tropismo a CXCR4, el patrón de resistencia a otros antirretrovirales es similar al de la población con tropismo a CCR5 en la basal, fundamentándose esto en los datos disponibles. De ahí que en la selección de un régimen de tratamiento, se debe asumir que los virus que forman parte de la población de CXCR4 que no se detectaron previamente (por ejemplo, la población viral menor), tiene el mismo patrón de resistencia que la población de virus con tropismo a CCR5. Fracaso con virus con tropismo a CCR5: Resistencia fenotípica: en pacientes con virus con tropismo a CCR5 al momento del fracaso del tratamiento con maraviroc, 22 de los 58 pacientes tenían virus con sensibilidad reducida al maraviroc. Además, los virus con tropismo a CCR5 de 2 de estos sujetos en los que falló el tratamiento tenían cambios ≥ 3 veces en los valores de la EC50 de maraviroc al momento del fracaso, pero la significancia de esto no es clara. En los pacientes restantes, no existió evidencia de una sensibilidad reducida según se identificaron por análisis virológicos exploratorios en un grupo representativo. Este último grupo tuvo marcadores de baja exposición, en algunos casos asociados con cumplimiento deficiente de tratamiento. Fracaso con virus usuarios de CXCR4: Se detectaron virus usuarios de CXCR4 en aproximadamente 28% (24/86) de sujetos con virus con tropismo CCR5 en la línea basal y en los que fracasó el tratamiento con maraviroc, en comparación con ninguno de los sujetos que experimentaron fracaso del tratamiento en el brazo de efavirenz. Con base en un re-análisis, cuando los sujetos que presentaban virus usuarios CXCR4 en la exploración, detectados usando un ensayo de tropismo con sensibilidad mejorada, fueron censurados del análisis, de los sujetos con virus con tropismo CCR5 en la línea basal y en los que fracasó el tratamiento con maraviroc, se detectaron virus usuarios de CXCR4 en 21% (25/118) en comparación con ninguno en el brazo de efavirenz. Se realizó un análisis clonal detallado en dos sujetos que no habían recibido tratamiento previo con antirretrovirales enrolados en un estudio con monoterapia Fase 2a y en los que se había observado virus usuarios CXCR4 después de 10 días de tratamiento con maraviroc. En forma consistente con el análisis clonal detallado realizado en sujetos que ya habían recibido tratamiento, se encontró que la variante de virus usuarios de CXCR4 existía antes del inicio de la terapia. Fracaso con virus con tropismo CCR5: Resistencia fenotípica: en pacientes con virus con tropismo CCR5 al momento del fracaso del tratamiento con maraviroc, 6 de 38 pacientes tenían virus con una sensibilidad reducida al maraviroc. En los 32 pacientes restantes, no había evidencia de virus con sensibilidad reducida lo cual se identificó mediante ensayos virológicos exploratorios en un grupo representativo. Un sujeto adicional presentaba un incremento de ≥3 veces en el valor CE50 para maraviroc al momento del fracaso. Estudios clínicos: Estudios en pacientes previamente tratados con VIH-1 con tropismo a CCR5: Se investigó la eficacia clínica de SELZENTRY® (en combinación con otros medicamentos antirretrovirales) en niveles plasmáticos del ARN del VIH y en los conteos de células CD4+ en dos estudios pivote, aleatorizados, doble ciego, multicéntricos, actualmente en curso (MOTIVATE 1 y MOTIVATE 2, n=1049) en pacientes infectados con VIH-1 CCR5 trópico. Los pacientes que fueron elegibles para estos estudios tuvieron una exposición previa a por lo menos 3 clases de medicamentos antirretrovirales, [≥1 ITRNs, ≥1 ITRNNs, ≥2 IPs, y/o enfuvirtida] o resistencia documentada a por lo menos un integrante de cada clase. Los pacientes fueron aleatorizados con una relación 2:2:1 para 300 mg de maraviroc (dosis equivalente) una vez al día, dos veces al día, o placebo, en combinación con una TBO, consistente en 3 a 6 medicamentos antirretrovirales (excluyendo a la dosis baja de ritonavir). La TBO se seleccionó con base en el historial previo del sujeto y las mediciones de resistencia viral genotípica y fenotípica.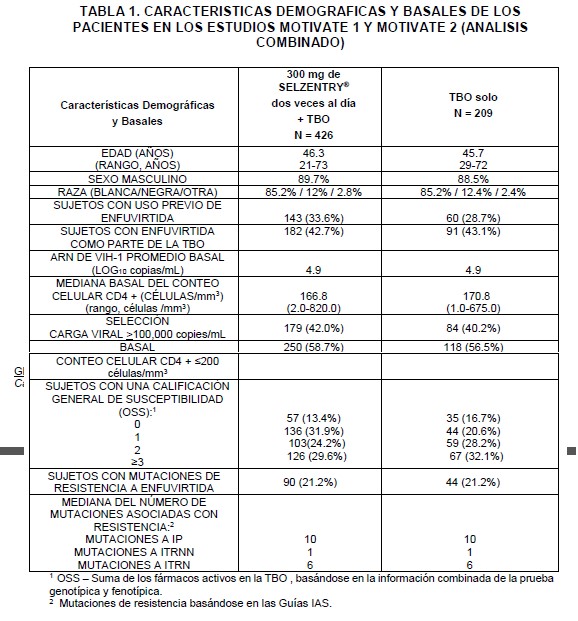
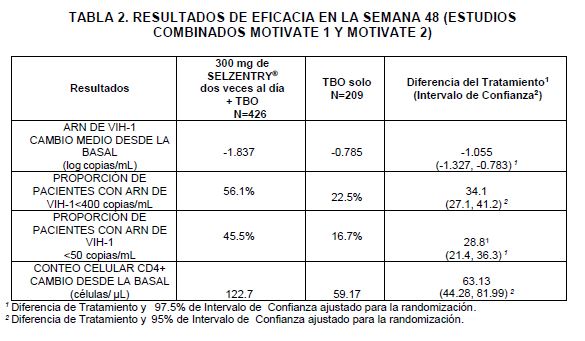
SELZENTRY® dos veces al día + TBO fueron superiores a la TBO solo, en los subgrupos de pacientes analizados (Ver Tabla 3).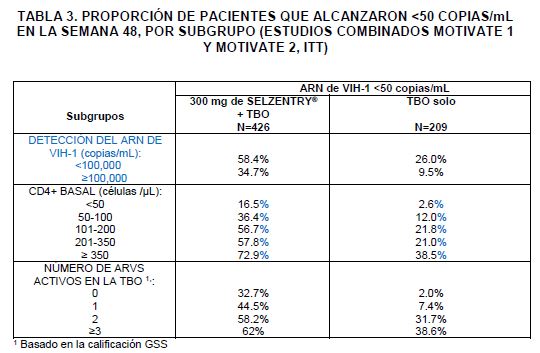
La determinación del tropismo para el enrolamiento a los estudios MOTIVATE se llevó a cabo mediante una prueba de tropismo fenotípico (Trofile). Esta fue reemplazada con una prueba de tropismo fenotípico más sensible (Trofile-ES), y se realizó un re-análisis retrospectivo de eficacia con esta prueba sólo en sujetos con virus R5 trópico. Los resultados de este análisis retrospectivo se presentan en la Tabla 4.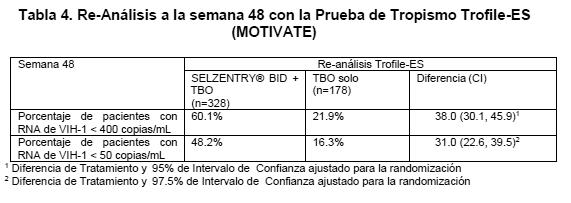
Estudios en Pacientes con VIH-1 sin tropismo para CCR5, tratados con anterioridad: El estudio A4001029 fue un estudio exploratorio, aleatorizado, doble ciego, multicéntrico para determinar la seguridad y eficacia de SELZENTRY® en sujetos infectados con el VIH-1 con tropismo a CXCR4 o dual/mixto. Los criterios de inclusión/exclusión fueron similares a los de los estudios MOTIVATE 1 y MOTIVATE 2 antes señalados y los sujetos fueron aleatorizados en una relación 1:1:1 al maraviroc una vez al día, maraviroc dos veces al día, o al placebo. En los sujetos que recibieron maraviroc, no se observó un riesgo acrecentado de infección, o una progresión de la enfermedad provocada por el VIH. El uso de maraviroc no se relacionó en estos sujetos, con una disminución significativa en el ARN del VIH-1, en comparación con el placebo, y no se notó un efecto adverso en el conteo de CD4. Estudios realizados en pacientes coinfectados con el virus de la Hepatitis B y/o Hepatitis C: Se evaluó la seguridad hepática de maraviroc en combinación con otros agentes antirretrovirales en sujetos infectados con VIH-1 con RNA de VIH < 50 copias/mL, coinfectados con el virus de la Hepatitis C y/o Hepatitis B en un estudio doble ciego, controlado con placebo, randomizado, multicéntrico. Se randomizó a 70 sujetos (Child-Pugh Clase A, n=64; Child-Pugh Clase B, n=6) en el grupo de maraviroc y se randomizó a 67 sujetos (Child-Pugh Clase A, n=59; Child-Pugh Clase B, n=8) en el grupo placebo. El objetivo primario evaluó la incidencia de anormalidades de la enzima hepática alanino aminotransferasa (ALT) grados 3 y 4, definidas por la elevación de 5 veces del límite superior normal (si los niveles basales de ALT estaban por debajo de ese límite) o por la elevación de 3.5 veces del límite superior normal (si los niveles basales de ALT estaban por encima de ese límite) en la semana 48. Un sujeto en cada brazo del tratamiento cumplió con el punto final primario para la semana 48 (en la semana 8 para el placebo y en la semana 36 para el brazo de maraviroc). Propiedades farmacocinéticas: Absorción: La absorción de maraviroc es variable con múltiples picos. La mediana de los picos de las concentraciones en plasma del maraviroc se consigue en 2 horas (rango 0.5 - 4 horas) posteriormente a las dosis orales únicas de la tableta comercial de 300 mg, administradas a voluntarios sanos. La farmacocinética del maraviroc oral no es proporcional a la dosis, en el rango de la dosis de 1-1200 mg. La biodisponibilidad absoluta de una dosis de 100 mg es de 23% y se predice que sea 33% con 300 mg. El maraviroc es un substrato para la salida de la glicoproteína P transportadora. La coadministración de una tableta de 300 mg con un desayuno alto en grasas, redujo la Cmax y el área bajo la curva (ABC) del maraviroc en un 33%, en voluntarios sanos. No existieron restricciones alimenticias en los estudios que demostraron la eficacia y seguridad del maraviroc (Ver Farmacodinamia). Por lo tanto el maraviroc se puede tomar con o sin alimentos, en las dosis recomendadas. Distribución: maraviroc se une (aproximadamente 76%) a las proteínas del plasma humano, y muestra una afinidad moderada por la albúmina y la glicoproteína alfa 1 ácida. El volumen de distribución del maraviroc es de aproximadamente 194 L. Metabolismo: Los estudios en humanos y los estudios in vitro utilizando microsomas hepáticos humanos y enzimas expresadas han demostrado que el maraviroc es metabolizado principalmente por enzimas del sistema citrocromo P450 hacia metabolitos que son esencialmente inactivos contra el VIH-1. Los estudios in vitro indican que la CYP3A4 es la enzima principal responsable del metabolismo del maraviroc. Los estudios in vitro también demostraron que las enzimas polimórficas CYP2C9, CYP2D6 y CYP2C19 no contribuyen significativamente al metabolismo del maraviroc. Maraviroc es el componente circulante principal (aproximadamente 42% de radioactividad) posterior a la administración de una dosis oral única de 300 mg. El metabolito circulante más significativo en humanos es una amina secundaria (aproximadamente 22% de radioactividad) formada por N-dealquilación. Este metabolito polar no presenta actividad farmacológica significativa. Otros metabolitos son productos de la mono-oxidación y son sólo componentes menores de la radioactividad del plasma. Eliminación: Se realizó un estudio de balance de masa / excreción utilizando una dosis única de 300 mg de maraviroc marcado con 14C. Aproximadamente el 20% del radioetiquetado se recupera en la orina y el 76% se recuperó en las heces en 168 horas. El maraviroc fue el mayor componente presente en la orina (promedio de 8% de la dosis) y en heces (promedio de 25% de la dosis). El resto fue excretado como metabolitos. Posteriormente a la administración intravenosa (30 mg), la vida media del maraviroc fue de 13.2 h, 22% de la dosis se excretó sin cambio en la orina y los valores de la depuración total y la depuración renal fueron de 44.0 L/h y 10.2 L/h, respectivamente. Poblaciones de pacientes especiales: Menores de edad: La farmacocinética del maraviroc en pacientes pediátricos menores de 18 años de edad no se ha establecido. Pacientes geriátricos: Se llevó a cabo un análisis de la población en los estudios Fase 1/2a y Fase 3 (16-65 años de edad) y no se observó un efecto de la edad. No se ha establecido la farmacocinética del maravicov en pacientes mayores de 65 años de edad (Ver Dosis y Vía de Administración). Insuficiencia renal: Un estudio comparó la farmacocinética de una dosis simple de 300mg de maraviroc en sujetos con insuficiencia renal severa (CLcr < 30mL / min, n = 6) y enfermedad renal en etapa terminal (ESRD) contra voluntarios sanos (n=6). La media geométrica del ABCinf (CV%) para maraviroc fue como sigue: voluntarios sanos (función renal normal) 1348.4 nanogramos·h/mL (61%); insuficiencia renal grave 4367.7 nanogramos·h/mL (52%); ESRD (dosificación después de la diálisis) 2677.4 nanogramos·h/mL (40%); y ESRD (dosificación antes de la diálisis) 2805.5 nanogramos·h/mL (45%). La C max (CV%) fue de 335.6 nanogramos/mL (87%) en los voluntarios sanos (función renal normal); 801.2 nanogramos/mL (56%) insuficiencia renal grave; 576.7 nanogramos/mL (51%) en ESRD (dosificación después de la diálisis) y 478.5 nanogramos/mL (38%) en ESRD (dosificación antes de la diálisis). La diálisis tuvo un efecto mínimo sobre la exposición en sujetos con ESRD. Las exposiciones observadas en sujetos con insuficiencia renal grave y con ESRD se encontraron dentro del intervalo de las observadas en estudios con dosis única de 300 mg de maraviroc en voluntarios sanos con función renal normal. Por lo tanto, no es necesario un ajuste de dosis en pacientes con insuficiencia renal recibiendo maraviroc sin un inhibidor potente de la CYP3A (Ver Dosis y Vía de Administración, Precauciones Generales e Interacciones). Adicionalmente, el estudio comparó la farmacocinética de dosis múltiples de maraviroc en combinación con saquinavir / ritonavir 1000/100 mg dos veces al día (una combinación inhibidora potente del CYP3A4) durante 7 días en sujetos con insuficiencia renal leve (CLcr > 50 y ≤80 mL / min, n = 6) e insuficiencia renal moderada (CLcr ≥30 y ≤50 mL / min, n = 6) a voluntarios sanos (n=6) Los sujetos recibieron 150 mg de maraviroc en diferentes frecuencias de dosis (voluntarios sanos - cada 12 horas; insuficiencia renal leve - cada 24 horas; insuficiencia renal moderada - cada 48 horas). La concentración promedio (Cavg) del maraviroc a lo largo de 24 horas fue de 445.1 nanogramos/mL, 338.3 nanogramos/mL, and 223.7 nanogramos/mL para sujetos con función renal normal, insuficiencia renal leve, e insuficiencia renal moderada, respectivamente. La Cavg del maraviroc de 24-48 horas para sujetos con insuficiencia renal moderada fue baja (Cavg: 32.8 nanogramos/mL). Por lo tanto, en sujetos con insuficiencia renal moderada (y por extrapolación en insuficiencia renal grave) la frecuencia de dosificación de más de 24 horas puede resultar en una exposición inadecuada entre las 24 y 48 horas. En pacientes con insuficiencia renal recibiendo maraviroc con inhibidores potentes de la CYP3A se recomienda una dosis de 150 mg cada 24 horas (Ver Dosis y Vía Administración, Precauciones Generales e Interacciones). Insuficiencia hepática: El maraviroc se metaboliza y elimina principalmente en el hígado. Un estudio comparó la farmacocinética de una dosis única de 300 mg de maraviroc en pacientes con insuficiencia hepática leve (Child-Pugh Clase A, n=8), y moderada (Child-Pugh Clase B, n=8), en comparación con sujetos sanos (n=8). Los índices de la media geométrica para la Cmax y ABCfinal fueron 11% y 25% más altos, respectivamente, en sujetos con insuficiencia hepática leve y 32% y 46% más altos para los sujetos con insuficiencia hepática moderada al compararse con sujetos con un funcionamiento hepático normal. Los efectos de la insuficiencia hepática moderada pueden subestimarse debido a los datos limitados sobre pacientes con capacidad metabólica disminuida y depuración renal mayor. Por lo tanto, los resultados deberán interpretarse con precaución. La farmacocinética del maraviroc no se ha estudiado en sujetos con insuficiencia hepática severa.(Ver Dosis y Vía Administración, Precauciones Generales). Raza: Los análisis farmacocinéticos de la población de los datos combinados Fase 1/2a, indicaron que la exposición fue 26.5% mayor en asiáticos (n=95), en comparación con no-asiáticos (n=318). Sin embargo, un estudio diseñado para evaluar las diferencias entre los caucásicos (n=12) y los asiáticos (n=12) no mostró una diferencia entre estas dos poblaciones. No es necesario un ajuste de dosis en base a la raza. Género: Los análisis farmacocinéticos de la población de los datos combinados de los estudios Fase 1/2a, indicaron que el género (mujeres: n=96, 23.2% de la población) no afecta las concentraciones del maraviroc. No se requiere un ajuste de dosis basándose en el género.
Contraindicaciones: Hipersensibilidad al principio activo o a cualquiera de los excipientes. Menores de 18 años de edad.
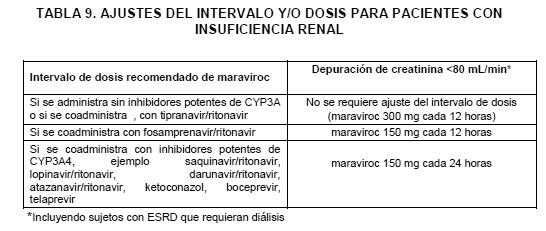
Precauciones generales: Tropismo: SELZENTRY® debe tomarse como parte de un régimen de combinación antirretroviral. SELZENTRY® deberá combinarse de forma óptima con otros antirretrovirales, a los cuales el virus del paciente sea sensible. (Ver Famacodinamía). SELZENTRY® únicamente debe utilizarse cuando sea detectable el tropismo para CCR5 (es decir cuando no se detecta el virus trópico CXCR4 o dual/mixto) detectado mediante una prueba de tropismo adecuadamente validada y sensible. (Ver Indicaciones, Dosis y Vía de Administración y Farmacodinamia). No es posible predecir el tropismo viral mediante el historial de tratamiento o el análisis de muestras almacenadas, solo puede usarse una muestra fresca del paciente para determinar el tropismo viral). Los cambios en el tropismo viral ocurren con el transcurso del tiempo en los pacientes infectados por el VIH-1. Por consiguiente, es necesario iniciar la terapia poco tiempo después de la prueba de tropismo. Ajuste de dosis: Los médicos deben asegurarse que se haga el ajuste de dosis adecuado de SELZENTRY®, cuando éste se co-administra con inhibidores y/o inductores de la CYP3A4, dado que las concentraciones de SELZENTRY® y sus efectos terapéuticos pueden afectarse. (Ver Dosis y Vía de Administración e Interacciones) Favor de remitirse también a la información del producto respectiva de los otros medicamentos antirretrovirales utilizados conjuntamente. Información para los pacientes: Se les debe advertir a los pacientes que las terapias antirretrovirales, incluyendo al SELZENTRY®, no han demostrado prevenir el riesgo de transmisión del VIH a otros a través del contacto sexual, o la contaminación con sangre. Los pacientes deben continuar tomando las precauciones adecuadas. Los pacientes también deberán estar informados de que SELZENTRY® no es cura para infección por VIH-1. Seguridad cardiovascular: Deberá utilizarse con cuidado en pacientes con un riesgo aumentado de eventos cardiovasculares. Durante los estudios Fase 3 en pacientes con VIH-1 con tropismo para CCR5 con tratamiento previo, diez sujetos (1.2%) que recibieron SELZENTRY® (en comparación con un bajo placebo), presentaron eventos de cardiopatías isquémicas [seis pacientes (1.4%) en el grupo de SELZENTRY® una vez al día y cuatro pacientes (0.9%) en el grupo de dos veces al día] Estos sujetos generalmente padecían enfermedad cardiaca o factores de riesgo cardiaco antes del uso de SELZENTRY®, y se desconoce la posible contribución del SELZENTRY® a dichos eventos. Hipotensión Postural: Cuando SELZENTRY® se administró en estudios con voluntarios sanos a dosis superiores que las recomendadas, se observaron con mayor frecuencia casos de hipotensión postural sintomática, en comparación con el placebo. Debera tenerse precaución cuando se administre SELZENTRY® a pacientes con insuficiencia renal grave, que tengan un historial de, o factores de riesgo para, hipotensión postural o que estén recibiendo medicamentos concomitantes que se sepa disminuyan la presión sanguínea. Pacientes con insuficiencia renal grave tratados con inhibidores potentes de la CYP3A o reforzados con inhibidores de la proteasa (PIs) tienen un mayor riesgo de presentar hipotensión postural debido al aumento en las concentraciones de maraviroc (véase Dosis y Administración, Interacciones y Farmacocinética). Pacientes con padecimientos cardiovasculares concomitantes pueden estar en un riesgo mayor de presentar eventos adversos cardiovasculares disparados por la hipotensión postural. Síndrome de reconstitución inmune: En pacientes infectados por VIH con inmunodeficiencia severa al momento de implementar una terapia antirretroviral altamente activa (TARAA), puede aparecer una reacción inflamatoria a patógenos oportunistas asintomáticos o residuales y provocar manifestaciones graves de una enfermedad, o bien la agravación de los síntomas. Generalmente, dichas reacciones se han observado durante las primeras semanas o meses de haber iniciado la terapia TARAA. Ejemplos relevantes son la retinitis por citomegalovirus, infecciones micobacterianas generalizadas y/o focalizadas, y la neumonía causada por Pneumocystis jiroveci (anteriormente conocida como Pneumocystis carinii). Cualquier síntoma inflamatorio debe ser evaluado y se debe iniciar tratamiento, cuando esto sea necesario. También se han reportado trastornos autoinmunes (tales como la enfermedad de Graves, polimiositis y síndrome de Guillain-Barre) ocurriendo en el síndrome inflamatorio de reconstitución inmune, sin embargo, el tiempo de presentación es más variable, y puede ocurrir muchos meses después del inicio del tratamiento y algunas veces puede ser de presentación atípica. Seguridad hepática: Se observó un incremento en las reacciones hepáticas adversas con SELZENTRY® durante los estudios de sujetos con infección por VIH tratados previamente, aunque no se presentó un incremento general en las anormalidades de las pruebas de funcionamiento hepático (ACTG Grado 3/4). (Ver Reacciones Adversas) Existieron menos casos de trastornos hepatobiliares reportados en pacientes que no habían recibido tratamiento previo y que se encontraban en tratamiento con SELZENTRY® que en pacientes que se encontraban bajo efavirenz pero la incidencia global de eventos adversos hepáticos y el Grado 3/4 para anormalidades en las pruebas de función hepática en pacientes que no habían recibido tratamiento previo fue similar entre SELZENTRY® y efavirenz. Se han reportado casos de hepatotoxicidad e insuficiencia hepática con características alérgicas en asociación con SELZENTRY®. Se debe considerar la descontinuación del SELZENTRY® en cualquier paciente con signos o síntomas de hepatitis aguda, en particular si se sospecha de hipersensibilidad relacionada con el fármaco, o de un incremento en las transaminasas hepáticas combinado con erupción cutánea u otros síntomas sistémicos de posible hipersensibilidad (por ejemplo, erupción cutánea pruriginosa, IgE elevada o eosinofilia). Existe información limitada en pacientes con co- infección de virus hepatitis B y/o C (véase Estudios Clínicos), deberá ejercerse precaución al tratar estos pacientes. En caso de terapia antiviral concomitante para la hepatitis B y/o C, favor de referirse a la información del producto pertinente para estos medicamentos. Los pacientes con una disfunción hepática pre-existente, incluyendo hepatitis activa crónica, pueden presentar un incremento en la frecuencia de las anormalidades en el funcionamiento hepático, durante la terapia antirretroviral combinada y deben ser monitoreados de acuerdo a la práctica estándar. La seguridad y eficacia de SELZENTRY® no ha sido estudiada específicamente en pacientes con trastornos hepáticos subyacentes significativos. Se cuenta con una limitada experiencia con pacientes con un funcionamiento hepático reducido, por lo tanto, SELZENTRY® debe utilizarse con precaución en esta población. Insuficiencia Renal: Un estudio evaluó la farmacocinética y seguridad de SELZENTRY® en pacientes con diferentes grados de insuficiencia renal comparados con voluntarios sanos. En este estudio, se observaron disminuciones transitorias en la CLcr media en sujetos con insuficiencia renal leve y moderada, así como en voluntarios sanos que recibieron SELZENTRY® 150 mg (Dosis frecuente: voluntarios sanos - una vez cada 12 horas; insuficiencia leve - una vez cada 24 horas; insuficiencia moderada - una vez cada 48 horas) y saquinavir/ritonavir 1000/100mg - dos veces al día la cual se resolvió con dosificación continua. No hubo relación entre las disminuciones en la CLcr media y la creatinina sérica basal media. Generalmente, SELZENTRY® fue bien tolerado en este estudio con más eventos adversos (mayormente leves) reportados en sujetos con insuficiencia renal leve y moderada que reciben SELZENTRY® y saquinavir/ritonavir. La tabla 9 proporciona normas para el ajuste de la dosis y/o intervalo para pacientes con insuficiencia renal con y sin inhibidores potentes de la CYP3A coadministrados. (Ver Dosis y Vía de Administración, Interacciones y Farmacocinética). Reacciones cutáneas y de hipersensibilidad graves: Se han reportado reacciones de hipersensibilidad incluyendo eventos graves y potencialmente riesgosos para la vida en pacientes tomando SELZENTRY®, en la mayoría de los casos en forma concomitante con otros fármacos asociados con estas reacciones. Estas reacciones estuvieron caracterizadas por aspectos que incluyeron rash, hallazgos constitucionales, y algunas veces disfunción orgánica e insuficiencia hepática. Se han reportado casos de síndrome de Stevens-Johnson (SJS), necrólisis epidérmica tóxica (NET) y rash medicamentoso con eosinofilia y síntomas sistémicos (DRESS por sus siglas en inglés) (referirse a Reacciones Adversas). Descontinúe el SELZENTRY® y otros agentes sospechosos inmediatamente si se desarrollan signos o síntomas de reacciones cutáneas o de hipersensibilidad graves. El retardo en el paro del tratamiento con SELZENTRY® o con otros fármacos sospechosos después del establecimiento del rash puede tener como resultado una reacción que ponga en riesgo la vida. Deberá monitorearse el estado clínico incluyendo a las aminotransferasas hepáticas y la terapia apropiada iniciada. Efectos sobre la capacidad para conducir y utilizar maquinaria: No se han realizado estudios sobre los efectos en la capacidad de conducir y utilizar maquinaria. Los pacientes deberán ser informados de que si experimentan mareo, deberán evitar tareas potencialmente riesgosas como conducir vehículos automotores u operar maquinaria. Restricciones de uso durante el embarazo y la lactancia: No existe información sobre los efectos del maraviroc sobre la fertilidad humana. En ratas, no existieron efectos adversos sobre fertilidad de machos o hembras (ver información preclínica). No se dispone de datos clínicos sobre la exposición durante el embarazo. Estudios en animales no mostraron efectos peligrosos directos o indirectos, en cuanto al embarazo, el desarrollo embrionario/fetal, del parto o postnatal. El maraviroc únicamente se debe utilizar durante el embarazo cuando el beneficio potencial justifique el posible riesgo del feto. Para evitar la transmisión del VIH, expertos en salud recomiendan que cuando sea posible las mujeres infectadas con VIH no amamanten a sus lactantes. Cuando se considere la leche materna durante el tratamiento antirretroviral, y donde no sea posible la alimentación con fórmula, deben seguirse las guías oficiales locales para la lactancia y el tratamiento. Aunque no se ha confirmado en humanos, y basándose en datos con animales, se espera que maraviroc sea secretado con la leche materna.
Reacciones secundarias y adversas: SELZENTRY® ha sido estudiado en 1374 pacientes infectados con VIH-1 multi-tratados, quienes recibieron por lo menos una dosis de maraviroc durante los estudios clínicos Fase 3. Esto incluye a 426 pacientes con tratamiento previo que recibieron la dosis recomendada de 300 mg dos veces al día y a 414 pacientes más, que recibieron 300 mg, una vez al día. El perfil de seguridad de SELZENTRY® se basa en 786 pacientes infectados con VIH -1 quienes recibieron 300mg de SELZENTRY® dos veces al día. La evaluación de las reacciones adversas relacionadas con el tratamiento se basó en los datos compilados de las dosis recomendadas para dos estudios Fase 3 en pacientes adultos a los que ya se les había administrado tratamiento (MOTIVATE 1 y MOTIVATE 2) en pacientes infectados con VIH-1 CCR5 trópico. Las reacciones adversas se listan por clase del sistema orgánico (CSO) y frecuencia. En cada grupo de frecuencia, las reacciones adversas se presentaron en un orden decreciente de gravedad. Las frecuencias se definieron como muy común (≥1/10), comunes (≥ 1/100 a < 1/10) y poco comunes (≥1/1000 a < 1/100). Las reacciones adversas y anormalidades de laboratorio que se presentan a continuación no están ajustadas a la exposición. Pacientes que ya habían recibido tratamiento: La Tabla 5 y Tabla 6 resumen toda la información del tratamiento doble ciego (dos veces al día=551, placebo=160 pacientes años de exposición) combinada a partir de los estudios Fase 3 MOTIVATE 1 y 2.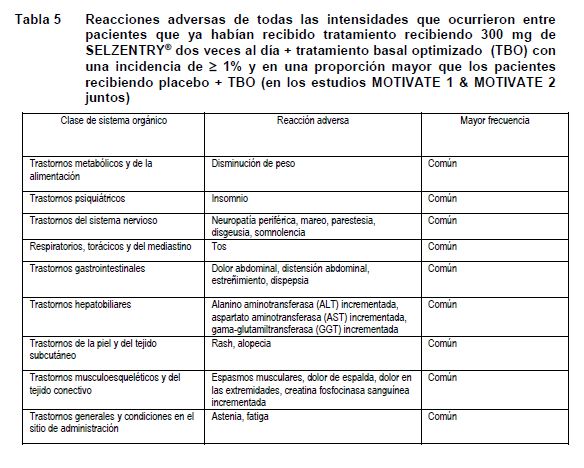
Anormalidades de laboratorio en pacientes que ya habían recibido tratamiento.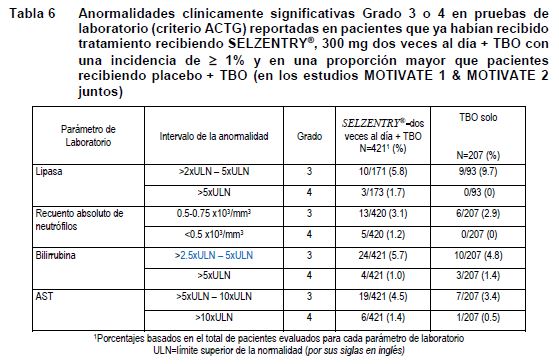
Los estudios MOTIVATE 1 y MOTIVATE 2 se abrieron después de la visita de la semana 48 del último paciente enrolado, así los pacientes elegibles pudieron cambiar a la extensión en fase abierta con MVC BID hasta la semana 96. Se completó una fase observacional subsecuente hasta los 5 años para evaluar la incidencia de Objetivos de Seguridad a Largo Plazo/Objetivos Selectos (LTS/SE por sus siglas en Inglés) incluyendo muerte, eventos definitorios de SIDA, insuficiencia hepática, IM/isquemia cardiaca, malignidades, rabdomiólisis y otros eventos infecciosos graves bajo tratamiento con MVC. La incidencia de esos objetivos selectos fue consistente con los datos de la semana 96. Información post comercialización: Muy ocasionalmente, se han reportado, reacciones de hipersensibilidad severas. Estas incluyen eritema medicamentoso con eosinofilos y síntomas sistémicos (DRESS), reacciones cutáneas severas (SJS y TEN) así como hepatotoxicidad e insuficiencia hepática con características alérgicas. En casos raros se han reportado hipotensión postural que pueden resultar en síncope.
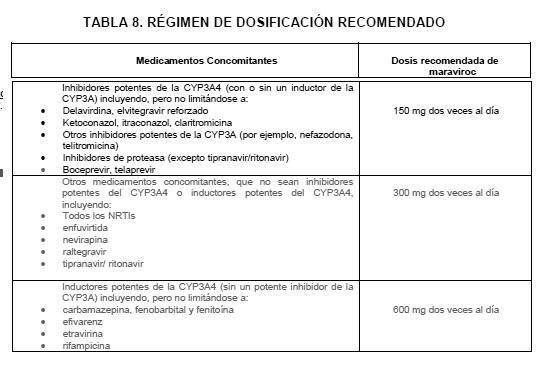
Interacciones medicamentosas y de otro género: El maraviroc es un substrato de la enzima CYP3A4 del sistema citocromo P450. La co-administración de SELZENTRY® con los medicamentos que inducen la CYP3A4 puede disminuir las concentraciones de maraviroc y reducir sus efectos terapéuticos. La co-administración de SELZENTRY® con medicamentos que inhiben a la CYP3A4 puede incrementar las concentraciones de SELZENTRY® en plasma. Se recomienda un ajuste de dosis del SELZENTRY® cuando se coadministra con los inhibidores y/o inductores de la CYP3A4. A continuación se proporcionan mayores detalles para la administración concomitante de medicamentos (Ver Tablas 7 y 8). Los estudios en sistemas recombinantes y de microsomas hepáticos humanos, han mostrado que el maraviroc no inhibe alguna de las enzimas P450 principales, en concentraciones clínicamente significativas (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4). SELZENTRY® no presenta efectos clínicamente relevantes en la farmacocinética del midazolam, los anticonceptivos orales etinilestradiol y levonorgestrel, o la relación cortisol/6b-hidroxicortisol urinario, lo que sugiere falta de inhibición o inducción de la CYP3A4 in vivo. A pesar de la falta de inhibición de CYP2D6 in vitro, maraviroc provocó un aumento en la proporción metabólica de debrisoquina a 600 mg QD, pero no a 300 mgdos veces al día. Por lo tanto a exposiciones más altas de SELZENTRY®, no se puede excluir una potencial inhibición de la CYP2D6. Basándose en los datos clínicos in vitro, es bajo el potencial del maraviroc para afectar la farmacocinética de medicamentos coadministrados. La depuración renal se considera de aproximadamente el 23% de la depuración total del SELZENTRY® cuando éste se administra sin los inhibidores CYP3A4. Dado que están involucrados ambos procesos, tanto el activo como el pasivo, existe la factibilidad de competencia en la eliminación de otras sustancias activas eliminadas por la vía renal. No obstante, la coadministración del maraviroc con el tenofovir (sustrato para la eliminación renal) y el Cotrimoxazol (que contiene trimetoprim, un inhibidor del transporte renal de cationes) no mostró efecto alguno en la farmacocinética del maraviroc. Adicionalmente, la coadministración de maraviroc con la lamivudina/zidovudina no mostró efecto alguno del SELZENTRY® en la farmacocinética de lamivudina (depurada principalmente por la vía renal) o de zidovudina (depuración renal y sin metabolismo por el P450). SELZENTRY® inhibe la P-glucoproteína in vitro (IC50 es 183 mM). Sin embargo maraviroc no afecta significativamente la farmacocinética de la digoxina in vivo, sugiriendo que SELZENTRY® no inhibe ni induce la actividad de la glicoproteína-P.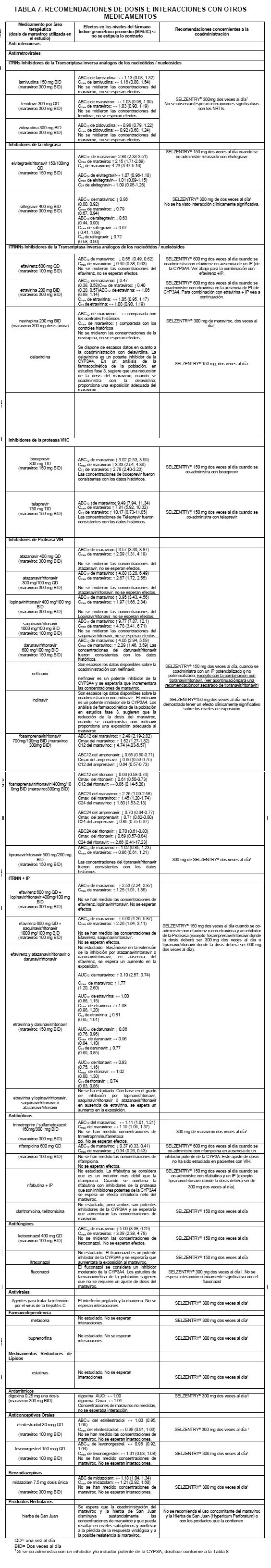
Alteraciones en los resultados de pruebas de laboratorio: La Tabla 6 muestra la incidencia ≥1% de las Anormalidades de Grado 3-4 (Criterios del ACTG) basadas en el cambio máximo en los valores de las pruebas de laboratorio independientemente de los valores basales en los estudios con tratamiento previo.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Información Preclínica: Los datos no clínicos no revelan algún peligro especial para los humanos con base en los estudios convencionales de seguridad farmacológica, toxicidad de dosis repetidas, genotoxicidad, carcinogenicidad potencial o de toxicidad en la reproducción. Carcinogénesis / mutagénesis: Se evaluó el potencial carcinogénico de maraviroc mediante un estudio de 6 meses en ratones transgénicos y un estudio de 24 meses en ratas. En ratones, el maraviroc no provocó un aumento estadísticamente significativo en la incidencia de algún tipo de tumor, a exposiciones sistémicas en un rango de 7 hasta 39 veces la exposición humana (basándose en la medición del ABC 0-24 horas no ligada) a una dosis máxima recomendada de 300 mg dos veces al día. En ratas la administración del maraviroc produjo adenomas tiroideos, relacionados con la adaptación de los cambios hepáticos a una exposición sistémica 21 veces la exposición humana de 300 mg de maraviroc dos veces al día. No se observó potencial carcinogénico para los humanos. El maraviroc no fue mutagénico o genotóxico en una batería de estudios in vitro e in vivo,