SERETIDE DISKUS
GSK
Denominación genérica: Salmeterol y Fluticasona
Forma farmacéutica y formulación: SERETIDE® DISKUS® Polvo. Cada dosis contiene: Xinafoato de Salmeterol equivalente a: 50mg 50md y 50mg de Salmeterol. Propionato de Fluticasona 100mg 250mg y 500mg. Excipiente cbp12.5mg.
Indicaciones terapéuticas: ASMA (Enfermedad Obstructiva Reversible de las vías aéreas): SERETIDE® DISKUS® está indicado para el tratamiento regular de asma (Enfermedad Obstructiva Reversible de la Vía Aérea). Esto puede incluir: Pacientes bajo dosis de mantenimiento efectivas de b2 agonistas de acción prolongada y corticoesteroides inhalados. Pacientes que continúan sintomáticos a pesar de estar recibiendo corticoesteroides inhalados. Pacientes tratados regularmente con broncodilatadores y que requieren adicionar corticoesteroides inhalados. Enfermedad Pulmonar obstructiva crónica (EPOC): SERETIDE® está indicado para el tratamiento regular de la Enfermedad Pulmonar Obstructiva Crónica (EPOC) incluyendo bronquitis crónica y enfisema, y ha demostrado que reduce la tasa de mortalidad por todas las causas.
Farmacocinética y farmacodinamia: Farmacocinética: No existe evidencia en animales y humanos en relación a que la administración concomitante por vía inhalada de Salmeterol y Fluticasona, afecte la farmacocinética de cada componente. Por lo tanto, para propósitos farmacocinéticos, cada componente puede ser considerado por separado. En estudios sobre interacciones medicamentosas, cruzado, con placebo y realizado en 15 sujetos sanos, la coadministración de SEREVENT®, (50 mcg administrados dos veces al día por inhalación) y el inhibidor del CYP3A4, ketoconazol (400mg administrados una vez al día vía oral), durante 7 días, produjo un aumento significativo en el grado de exposición plasmática al Salmeterol (1.4 veces la Cmax y 15 veces el AUC). No hubo aumento alguno en la acumulación de Salmeterol cuando se administraron dosis repetidas. Tres sujetos fueron retirados del tratamiento concomitante con SEREVENT® y ketoconazol debido a que experimentaron una prolongación en el intervalo QTc, o palpitaciones con taquicardia sinusal. En los 12 sujetos restantes la coadministración de SEREVENT® y ketoconazol no produjo efectos clínicamente significativos en la frecuencia cardiaca, el potasio sanguíneo o la duración del intervalo QTc (Véase en Precauciones Generales). Salmeterol: Actúa localmente a nivel pulmonar, por lo tanto, las concentraciones plasmáticas no son indicadores de sus efectos terapéuticos. Además, existe información limitada de la farmacocinética de Salmeterol debido a la dificultad técnica para analizar el fármaco en el plasma por sus bajas concentraciones después de la administración de dosis terapéuticas por vía inhalada (aproximadamente 200 picogramos/mL o menores). Después de la dosificación regular con Xinafoato de Salmeterol, puede detectarse ácido hidroxinaftóico en la circulación sistémica, el cual alcanza concentraciones en estado estable de aproximadamente 100 nanogramos/mL. Estas concentraciones son hasta 1000 veces más bajas que los niveles en estado estable que se observan en los estudios de toxicidad. No se han observado efectos perjudiciales después de la administración regular de Salmeterol a largo plazo (más de 12 meses) en pacientes con obstrucción de la vía aérea. En un estudio in vitro, se demostró que el salmeterol se metaboliza ampliamente a a-hidroxisalmeterol (oxidación alifática), a través de la isoenzima 3A4 del citocromo P450 (CYP3A4). Sin embargo, en un estudio realizado en voluntarios sanos que recibieron dosis repetidas de salmeterol y eritromicina, no se observaron cambios clínicamente significativos en los efectos farmacodinámicos, al administrar un régimen posológico de 500 mg de eritromicina tres veces al día. Sin embargo, en un estudio sobre interacciones de Salmeterol-Ketoconazol ocurrió un aumento significativo en el grado de exposición plasmática al salmeterol. (Véase en Precauciones Generales e Interacciones Medicamentosas y de otro Género). Propionato de Fluticasona: La biodisponibilidad absoluta del Propionato de Fluticasona en cada uno de los dispositivos inhaladores disponibles ha sido estimada a partir de las comparaciones realizadas en y entre los estudios de datos farmacocinéticos de las formulaciones inhalada e intravenosa. En sujetos adultos sanos, se ha estimado la biodisponibilidad absoluta para el propionato de fluticasona en Accuhaler/Diskus (7.8%), el propionato de fluticasona en Diskhaler (9.0%), el propionato de fluticasona Evohaler (10.9%), la combinación de sameterol-propionato de flutocasona en Evohaler (5.3%) y la combinación de salmeterol- propionato de fluticasona en Accuhaler /Diskus (5.5%), respectivamente. En pacientes que padecen asma o EPOC, se ha observado un menor grado de exposición sistémica al propionato de fluticasona. La absorción ocurre principalmente a nivel pulmonar y es inicialmente rápida, posteriormente prolongada. El resto de la dosis inhalada puede ser deglutida, sin embargo, su contribución es mínima para que haya una exposición sistémica debido a su baja solubilidad acuosa y metabolismo presistémico, resultando en una biodisponibilidad oral menor del 1%. Hay un incremento lineal en la exposición sistémica con el incremento de la dosis inhalada. La disposición del Propionato de Fluticasona se caracteriza por una alta depuración plasmática (1,150 mL/min), un amplio volumen de distribución en estado estable (aproximadamente de 300 L) y una vida media terminal de aproximadamente 8 h. La fijación a proteínas plasmáticas es moderadamente alta (91%). El Propionato de Fluticasona es eliminado rápidamente de la circulación sistémica principalmente por la vía metabólica a un metabolito inactivo: ácido carboxílico, a través de la enzima CYP3A4 del citocromo P450. La depuración renal de Propionato de Fluticasona es insignificante ( < 0.2%) y menos del 5% como el metabolito. Debe de tenerse cuidado cuando se coadministren inhibidores de la CYP3A4 conocidos, ya que existe un riesgo potencial de aumento a la exposición sistémica a Propionato de Fluticasona. Poblaciones de Pacientes Especiales: El análisis farmacocinético poblacional se realizó utilizando los datos obtenidos en sujetos asmáticos (nueve estudios clínicos realizados con PF y cinco estudios clínicos realizados con salmeterol) y demostró lo siguiente: Después de administrar SERETIDE® (50/100 microgramos), se observó un mayor grado de exposición al PF, en comparación con la administración de PF solo (100 microgramos), en adolescentes y adultos (relación de 1.52 [IC del 90% 1.08, 2.13]) y niños (relación de 1.20 [IC del 90% 1.06, 1.37]). En los niños que tomaron SERETIDE® (50/100 microgramos), se observó un mayor grado de exposición al PF, en comparación con los adolescentes y adultos (relación de 1.63 [IC del 90% 1.35, 1.96]). Se desconoce la importancia clínica de estos hallazgos; sin embargo, no se observaron diferencias en los efectos producidos en el eje HHS en los estudios clínicos de hasta 12 semanas de duración, en los cuales se comparó SERETIDE (50/100 microgramos) y PF (100 microgramos), tanto en adolescentes y adultos como en niños. Al administrar la dosis más alta de SERETIDE (50/500 microgramos), el grado de exposición al PF fue similar al observado con la dosis equivalente de PF solo. En los niños que tomaron SERETIDE® (50/100 microgramos), se observó un mayor grado de exposición al salmeterol, en comparación con los adolescentes y adultos (relación de 1.23 [IC del 90% 1.10, 1.38]). Se desconoce la importancia clínica de estos hallazgos; sin embargo, no se observaron diferencias en los efectos cardiovasculares o reportes de temblores, entre adultos, adolescentes y niños, en los estudios clínicos de hasta 12 semanas de duración. Farmacodinamia: Estudios clínicos realizados con SEREVENT: Asma: El estudio multricéntrico sobre Asma realizado con Salmeterol (SMART por sus siglas en Inglés [Salmeterol Multi-center Asthma Research Trial]) fue un estudio a gran escala realizado en los Estados Unidos en el que se comparó la seguridad de SEREVENT® y la de placebo como terapia adicional a la terapia usual del paciente. En este estudio no se detectaron diferencias significativas en el criterio principal de valoración que consistió en la combinación del número de muertes relacionadas con problemas respiratorios y número de experiencias relacionadas con el aparato respiratorio que pusieron en riesgo la vida del paciente. Este estudio mostro un incremento significativo en el número de muertes relacionadas con el asma en el grupo de pacientes que se encontraban recibiendo SEREVENT® (13 fallecimientos 13,176 pacientes tratados con SEREVENT® por 28 semanas contra 3 fallecimientos de 13,179 pacientes tratados con placebo). El estudio no fue diseñado para valorar el impacto del uso concurrente con una terapia con corticoesteroides inhalados. Sin embargo, en los análisis post-hoc se demostró que no existió una diferencia significativa entre los grupos de tratamiento en cuanto a las muertes relacionadas con el asma para aquellos pacientes que utilizaban corticoesteroides inhalados en la línea basal (4/6127 pacientes en el grupo de SEREVENT® contra 3/6138 pacientes con el grupo de placebo). El número de muertes relacionadas con el asma en aquellos grupos que no usaban corticoesteroides inhalados fue de 9/7049 pacientes en el grupo de SEREVENT® contra 0/7041 pacientes en el grupo de placebo. Adicionalmente, en una meta análisis de estudios de 42 estudios clínicos involucraron un total de 8,030 pacientes bajo tratamiento con SERETIDE® y 7,925 pacientes bajo tratamiento con FLIXOTIDE® no se demostró una diferencia significativa entre SERETIDE® y FLIXOTIDE® en cuanto a la incidencia de eventos graves del aparato respiratorio o en la incidencia de hospitalizaciones debidas a eventos asmáticos. Estudios clínicos con SERETIDE®: Asma: Un estudio de doce meses, en gran escala (obteniendo un Control Óptimo del Asma [Gaining Optimal Asthma ControL, GOAL]), en 3416 pacientes asmáticos comparó la eficacia y seguridad de SERETIDE® contra la monoterapia con el corticoesteroide inhalado para lograr los niveles predefinidos de control del asma. El tratamiento se llevó a cabo en etapas escalonadas cada 12 semanas hasta que se lograba el 'control total'** o se llegaba a la máxima dosis del fármaco en estudio. El control debía mantenerse durante por lo menos 7 de las últimas 8 semanas de tratamiento. El estudio demostró que: 71% de los pacientes tratados con SERETIDE® lograron un asma "bien controlada" en comparación con 59% de los pacientes tratados solamente con el corticoesteroide inhalado. 41% de los pacientes tratados con SERETIDE® lograron el "control total"** del asma en comparación con 28% de los pacientes tratados solamente con el corticoesteroide inhalado. Estos efectos se observaron en una etapa más temprana con SERETIDE® (y con una dosis más baja del corticoesteroide inhalado), en comparación con el corticoesteroide inhalado solo. El estudio GOAL también demostró que: La frecuencia de exacerbaciones con SERETIDE® fue 29% más baja en comparación con la monoterapia con el corticoesteroide inhalado. El logro de un asma "bien controlada" y "totalmente controlada" mejoró la Calidad de Vida (CdV). Después del tratamiento con SERETIDE®, 61% de los pacientes reportaron deterioro mínimo o ningún deterioro de la CdV, medida por un cuestionario específico para determinar la calidad de vida en los pacientes con asma, en comparación con 8% en la evaluación basal. *Asma bien controlada; 2 días o menos con síntomas de valor mayor a 1 (definiendo síntomas con valor de 1 como 'síntomas durante un corto periodo durante el día'), o uso de beta2 agonistas de corto acción (ABCDA) durante menos de o hasta 2 días, y menos de o hasta en 4 ocasiones/semana, 80% del valor predicho del volumen espiratorio máximo matutino, sin despertares nocturnos, sin exacerbaciones y sin efectos secundarios que obliguen a cambiar el tratamiento. ** Control total del asma; sin síntomas, sin uso de beta agonistas de acción corta, o igual o más del 80% o más del valor predicho del volumen espiratorio máximo, sin despertares nocturnos, sin exacerbaciones y sin efectos secundarios que obliguen a cambiar el tratamiento. Otros dos estudios han mostrado mejorías de la función pulmonar, el porcentaje de días sin síntomas y la reducción del uso de la medicación de rescate, una dosis del corticoesteroide inhalado 60% más baja con SERETIDE® en comparación con la monoterapia con el corticoesteroide inhalado, al mismo tiempo que se mantuvo el control de la inflamación subyacente de la vía aérea, medida por biopsia bronquial y lavado bronqueoalveolar. Otros estudios han demostrado que el tratamiento con SERETIDE® mejora significativamente los síntomas del asma y la función pulmonar, y reduce el uso de la medicación de rescate en comparación con el tratamiento con los componentes individuales por sí solos y con el placebo. Los resultados del estudio GOAL demuestran que las mejorías observadas con SERETIDE® en estos puntos finales se mantienen durante por lo menos 12 meses. Epoc: Pacientes sintomáticos sin restricción con una reversibilidad de 10% a un b2 agonista de acción corta: Pruebas clínicas controladas con placebo durante 6 meses han demostrado que el uso regular de SERETIDE® 50/250 y 50/500 mg, mejora rápida y significativamente la función pulmonar y reduce en grado significativo la disnea y el uso de medicamentos de rescate. También hubo una mejoría significativa en el estado de salud. Pacientes sintomáticos con EPOC que demostraron una reversibilidad menor a 10% a un b2-agonista de acción corta: Estudios clínicos controlados con placebo durante un periodo de 6 y 12 meses han demostrado que el uso regular de SERETIDE® 50/500 mg, mejora rápida y significativamente la función pulmonar y reduce en grado significativo la disnea y el uso de medicamentos de rescate. Durante un periodo de más de 12 meses disminuyó notablemente el riesgo de exacerbaciones de la EPOC y la necesidad de administrar cursos adicionales de corticoesteroides orales. También hubo una mejoría significativa en el estado de salud. SERETIDE® 50/500 mg fue eficaz para mejorar la función pulmonar y el estado de salud, así como para reducir el riesgo de exacerbaciones de la EPOC, tanto en fumadores, como en ex fumadores. Estudio TORCH (TOwards a Revolution in COPD Health [Hacia una Revolución en la Salud de los Pacientes con EPOC]): El estudio TORCH fue un estudio de 3 años de duración que se realizó para evaluar el efecto que ejerce el tratamiento con SERETIDE® DISKUS® 50/500 mg dos veces al día, comparado con 50 mg de salmeterol en DISKUS® dos veces al día, 500 mg de Propionato de Fluticasona (PF) en DISKUS® dos veces al día, o placebo, en la tasa de mortalidad por todas las causas, en pacientes que padecen EPOC. Aquellos pacientes que exhibieron una EPOC de grado moderado a severo, con un VEF1 en la línea basal (antes de utilizar algún broncodilatador) < 60% del valor normal predicho, fueron distribuidos aleatoriamente para recibir un tratamiento con medicamento doblemente ciego. Durante el estudio, se permitió que los pacientes recibieran un tratamiento ordinario de la EPOC, a excepción de otros corticoesteroides inhalados, broncodilatadores de acción prolongada y corticoesteroides sistémicos a largo plazo. En todos los pacientes se determinó es estado de supervivencia a los 3 años, independientemente del retiro de la medicación del estudio. El criterio principal de la valoración fue la reducción en la tasa de mortalidad por todas las causas, a los 3 años de tratamiento con SERETIDE® frente a placebo.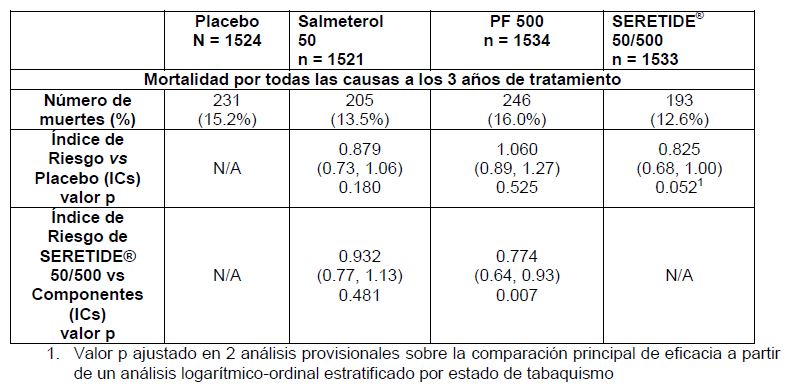
Durante los 3 años de tratamiento, SERETIDE® redujo el riesgo de muerte en cualquier momento en 17.5%, en comparación con el placebo (Índice de Riesgo de 0.825 (IC del 95%: 0.68, 1.00, p=0.052; todos ajustados en análisis provisionales). En el grupo tratado con salmeterol, hubo una reducción de 12% en el riesgo de muerte en cualquier momento, y por cualquier causa, durante los 3 años de tratamiento, en comparación con el grupo que recibió tratamiento con placebo (p=0.180), así como un aumento de 6% en el grupo tratado con PF en comparación con el placebo (p=0.525). Un análisis complementario que hizo uso del modelo de Riesgos Proporcionales de Cox arrojó un índice de riesgo de 0.811 (IC del 95%: 0.670, 0.982, p=0.031) en la comparación de SERETIDE® frente al placebo, lo cual representa una reducción de 19% en el riesgo de muerte en cualquier momento durante los 3 años de tratamiento. El modelo se ajustó a factores importantes (estado de tabaquismo, edad, género, región, VEF1 basal e índice de Masa Corporal). No hubo indicios de variaciones, ocasionadas por estos factores, en los efectos del tratamiento. El porcentaje de pacientes que murieron durante los 3 años de tratamiento debido a causas relacionadas con la EPOC, fue de 6.0% en el grupo tratado con placebo, 6.1% en el grupo tratado con salmeterol, 6.9% en el grupo tratado con PF y 4.7% en el grupo tratado con SERETIDE®. SERETIDE® redujo la tasa de exacerbaciones de grado moderado a severo en 25% (IC del 95%: 19% a 31%; p < 0.001), en comparación con el placebo. SERETIDE® redujo la tasa de exacerbaciones en 12%, en comparación con el salmeterol (IC del 95%: 5% a 19%, p=0.002), y 9% en comparación con el salmeterol (IC del 95%: 5% a 19%, p=0.002), y 9% en comparación con el PF (IC del 95%: 1% a 16%, p=0.024). El salmeterol y el PF redujeron significativamente las tasas de exacerbaciones en comparación con el placebo; es decir, en un 15% (IC del 95%: 7% a 22%; p < 0.001) y 18% (IC del 95%: 11% a 24%; p < 0.001), respectivamente. La Calidad de Vida Relacionada con la Salud, cuantificada a través del Cuestionario Respiratorio de St. George (SGRQ, por sus siglas en inglés), experimentó una mejoría con todos los tratamientos activos, en comparación con el placebo. El promedio de la mejoría observada durante los tres años de tratamiento con SERETIDE®, fue de -3.1 unidades en comparación con el placebo, (IC del 95%: - 4.1 a 2.1; p < 0.001), de -2.2 unidades (p < 0.001) en comparación con el salmeterol y de -1.2 unidades (p=0.017) en comparación con el PF. Durante el periodo de tratamiento de 3 años de duración, los valores de VEF1 fueron superiores en los sujetos tratados con SERETIDE® que en los que recibieron placebo (diferencia promedio durante los 3 años de tratamiento de 92 mL, IC del 95%: 75 a 108 mL; p < 0.001). Además, SERETIDE® fue más eficaz que el salmeterol o el PF en mejorar los valores de VEF1 (diferencia promedio de 50 mL, p < 0.001 en el grupo tratado con salmeterol y 44 mL, p < 0.001 en el grupo tratado con PF). La probabilidad estimada durante los 3 años de padecer neumonía notificada como un efecto adverso fue de 12.3% en el grupo tratado con placebo, 13.3% en el grupo tratado con salmeterol, 18.3% en el grupo tratado con PF y 19.6% en el grupo tratado con SERETIDE® (Índice de riesgo en la comparación SERETIDE® frente a placebo: 1.64, IC del 95%: 1.33 a 2.01, p < 0.001). No hubo aumento alguno en la tasa de muerte relacionada con neumonía; las muertes ocurridas durante el tratamiento que se adjudicaron como relacionadas principalmente con la neumonía fueron 7 en el grupo tratado con placebo, 9 en el grupo tratado con salmeterol, 13 en el grupo tratado con PF y 8 en el grupo tratado con SERETIDE®. No hubo diferencias significativas en la probabilidad de ocurrencia de fracturas óseas (5.1% en el grupo que recibió placebo, 5.1% en el grupo que recibió salmeterol, 5.4% en el grupo que recibió PF y 6.3% en el grupo que recibió SERETIDE®; Índice de riesgo en la comparación SERETIDE® frente a placebo: 1.22, IC del 95%: 0.87 a 1.72, p=0.248). La tasa de incidencia de efectos adversos de trastornos oculares, trastornos óseos y trastornos en el eje HHS fue baja y, además, no se observó diferencia alguna entre los tratamientos. En los grupos de tratamiento que recibieron salmeterol, no hubo indicios de aumentos en la tasa de incidencia de efectos adversos cardiacos. Mecanismo de acción: SERETIDE® DISKUS® contiene Salmeterol y Propionato de Fluticasona, los cuales, tienen diferente mecanismo de acción. Salmeterol protege contra los síntomas; Propionato de Fluticasona mejora la función pulmonar y previene las exacerbaciones. SERETIDE® DISKUS® puede ofrecer un régimen más conveniente para los pacientes bajo terapia combinada con un b-agonista y un corticoesteroide inhalados. Los mecanismos de acción de ambos fármacos se exponen a continuación. Salmeterol: Es un b2 -agonista selectivo de acción prolongada (12 horas), el cual posee una larga cadena lateral la cual se une al exo-sitio del receptor. Estas propiedades farmacológicas del Salmeterol ofrecen una mayor protección contra la broncoconstricción inducida por la histamina y producen una broncodilatación de mayor duración, por lo menos de 12 horas, que las dosis recomendadas de b2-agonistas convencionales de acción corta. Diversas pruebas in vitro han demostrado que Salmeterol es un inhibidor potente y de larga duración, en el pulmón humano, de mediadores químicos liberados de los mastocitos como son la histamina, los leucotrienos y prostaglandina D2. En el ser humano, Salmeterol inhibe las fases temprana y tardía de la respuesta a un alérgeno inhalado; esta última con una persistencia de más de 30 horas después de una sola dosis cuando el efecto broncodilatador ya no es evidente. La administración de dosis únicas de Salmeterol atenúa la hiperreactividad bronquial. Estas propiedades indican que el Salmeterol tiene un efecto adicional no broncodilatador, pero el significado clínico completo aún no es claro. Este mecanismo es diferente de los efectos antiinflamatorios de los corticoesteroides. Propionato de Fluticasona: El propionato de Fluticasona por inhalación en las dosis recomendadas, tiene una potente acción antiinflamatoria glucocorticoide a nivel pulmonar, resultando en una reducción de síntomas y exacerbaciones del asma, sin los efectos secundarios de los corticoesteroides administrados por vía sistémica. La producción diaria de hormonas corticosuprarrenales permanece dentro de los límites normales durante el tratamiento prolongado con Propionato de Fluticasona administrado por vía inhalada, aún, a la máxima dosis recomendada en niños y/o adultos. Después de la transferencia de otros esteroides inhalados, la producción diaria mejora gradualmente, a pesar del uso pasado y presente intermitente de esteroides orales, con lo que demuestra el retorno de la función suprarrenal normal bajo Propionato de Fluticasona inhalado. La reserva suprarrenal también permanece normal durante el tratamiento crónico, según se mide por el incremento normal en una prueba de estimulación. Sin embargo, es posible que durante un tiempo considerable persista algún deterioro residual de la reserva suprarrenal por un tratamiento previo y debe tenerse presente. (Véase Precauciones Generales)
Contraindicaciones: Pacientes con antecedentes de hipersensibilidad a cualquier componente de la fórmula.
Precauciones generales: SERETIDE® DISKUS® no debe utilizarse para el alivio de los síntomas agudos, para ello, se requiere de un broncodilatador de acción rápida y corta duración (ejemplo: salbutamol). Los pacientes deben de ser informados para que lleven consigo este tratamiento todo el tiempo. El incremento en el uso de broncodilatadores de acción corta para aliviar los síntomas, indica deterioro del control del asma y los pacientes deben ser examinados por un médico. Un deterioro repentino y progresivo del control del asma puede amenazar la vida del paciente, por lo tanto, deberá ser evaluado por un médico. Debe de considerarse la posibilidad de incrementar la terapia con corticoesteroides. Adicionalmente, si la posología manejada de SERETIDE® DISKUS® no ha permitido un control adecuado de la enfermedad, el paciente deberá ser evaluado nuevamente por un médico. En los pacientes con asma el tratamiento con SERETIDE® DISKUS® no debe suspenderse en forma abrupta, debido al riesgo de exacerbación; el tratamiento debe ser descontinuado en forma gradual, bajo supervisión médica. En los pacientes con EPOC, la suspensión del tratamiento puede asociarse con descompensación sintomática y debe de ser supervisada por un médico. En los estudios realizados en pacientes con EPOC, que recibieron tratamiento con SERETIDE®, se produjo un aumento en la tasa de notificaciones de neumonía (véase Reacciones secundarias y adversas). Se recomienda a los médicos instituir una vigilancia continua para evitar el posible desarrollo de neumonía en la EPOC, ya que las características clínicas de la neumonía suelen traslaparse con la exacerbación de la enfermedad. Como sucede con todos los fármacos inhalados que contienen corticoesteroides, SERETIDE® debe administrarse con precaución en pacientes con tuberculosis pulmonar activa o latente. SERETIDE® debe administrarse con precaución en pacientes con tirotoxicosis. En algunas ocasiones se pueden observar efectos cardiovasculares, como elevación de la tensión arterial sistólica y de la frecuencia cardiaca, con todos los fármacos simpaticomiméticos, especialmente cuando se administran a dosis superiores de las terapéuticas. Por esta razón, SERETIDE® debe administrarse con precaución a los pacientes con enfermedades cardiovasculares preexistentes. Existe la posibilidad de que se produzca un descenso transitorio en las concentraciones séricas de potasio con todos los fármacos simpaticomiméticos que se administran a dosis más altas que las terapéuticas. Por lo tanto, SERETIDE® debe administrarse con precaución a los pacientes que exhiben una predisposición a presentar bajas concentraciones séricas de potasio. Diversos efectos sistémicos pueden llegar a presentarse con el uso de cualquier corticoesteroide administrado por vía inhalada, particularmente cuando se manejan dosis altas durante periodos prolongados; hay una probabilidad mucho mayor de que estos efectos se produzcan al administrar corticoesteroides por vía oral (ver Sobredosis). Los posibles efectos incluyen: Síndrome de Cushing, rasgos cushingoides, supresión adrenal, retardo en el crecimiento en niños y adolescentes, disminución de la densidad mineral ósea, cataratas y glaucoma. Por lo tanto, es importante que para los pacientes con asma, que la dosis de los corticoesteroides inhalados sea titulada a la dosis más baja, con la cual se obtenga un control eficaz. La posibilidad del deterioro de la respuesta adrenal, siempre debe tenerse presente en las situaciones de urgencia y electivas con probabilidad de producir estrés y se debe considerar el tratamiento apropiado con corticoesteroides (ver sección Sobredosis). Se recomienda que en niños que reciban tratamiento prolongado con corticoesteroides inhalados, la estatura sea monitoreada regularmente. Debido a la posibilidad de presentarse una respuesta adrenal alterada, deberá tenerse especial precaución con los pacientes que sean transferidos de la terapia esteroidea oral a terapia con Propionato de Fluticasona inhalado, y la función corticosuprarrenal debe ser monitoreada regularmente. Posterior a la introducción de Propionato de Fluticasona inhalado, la suspensión de la terapia sistémica deberá ser gradual y se debe recomendar a los pacientes llevar consigo una tarjeta de precaución que indique la posible necesidad de recibir terapia esteroidea adicional en situaciones de estrés. En muy raras ocasiones, ha habido reportes de incremento en las concentraciones de glucosa en sangre (ver sección Reacciones Adversas); esto debe ser tomado en cuenta cuando se prescriba SERETIDE® a pacientes con antecedentes de diabetes mellitus. Durante su uso posterior a la comercialización, ha habido reportes de interacciones medicamentosas clínicamente significativas en pacientes que reciben propionato de fluticasona y ritonavir, dando como resultado efectos relacionados con la administración de corticoesteroides sistématicos, incluyendo síndrome de Cushing y supresión suprarrenal. Por tanto, se debe evitar el uso concomitante de propionato de fluticasona y ritonavir, a menos que el beneficio potencial para el paciente exceda el riesgo de experimentar efectos secundarios relacionados con la administración de corticoesteroides sistémicos. (Véase en Interacciones medicamentosas y de otro género). La información proveniente de un estudio a gran escala realizado en Estados Unidos (SMART), el cual comparó la seguridad de SEREVENT® (un componente de SERETIDE®) o de placebo, cuando se añadieron a la terapia regular del paciente, mostró un incremento significativo en el número de muertes relacionadas con el asma en los pacientes recibiendo SEREVENT®. La información de este estudio, sugirió que los pacientes afro-americanos, podrían encontrarse bajo un riesgo mayor de desarrollar eventos respiratorios graves o muerte al usar SEREVENT®, comparado con placebo. El estudio SMART no fue diseñado para determinar si el uso concurrente de corticoesteroides inhalados modifica el riesgo de experimentar muerte relacionada con el asma. En un estudio sobre interacciones medicamentosas, se observo que el uso concomitante de ketoconazol sistémico incrementa el grado de exposición a SEREVENT®. Esto podría ocasionar una prolongación en el intervalo QTc. Se debe tener cuidado al coadministrar potentes inhibidores de la isoenzima CYP3A4. (p.ej., Ketoconazol) con SEREVENT®. (Véase Interacciones medicamentosas y de otro género) Como ocurre con otras terapias inhaladas puede ocurrir broncoespasmo paradójico con un aumento inmediato de las sibilancias después de la dosificación. Esto debe tratarse inmediatamente con un broncodilatador inhalado de acción rápida y corto. Salmeterol-FP DISKUS® o Evohaler® deben descontinuarse inmediatamente, se debe evaluar al paciente e instituir una terapia alternativa si fuera necesario. (Véase Reacciones Secundarias y Adversas) Se han reportado reacciones adversas del tratamiento farmacológico con agonistas beta-2, tales como temblor, palpitaciones subjetivas y cefalea, pero tienden a ser transitorias y a disminuir con la regularización de la terapia. (véase Reacciones Secundarias y Adversas). Efectos en la Capacidad de Conducir y Operar Maquinaria: No se han realizado estudios específicos para evaluar el efecto que ejerce SERETIDE® DISKUS® en las actividades arriba mencionadas, pero el perfil farmacológico de ambos fármacos no indica la existencia de algún efecto.
Restricciones de uso durante el embarazo y la lactancia: La administración de fármacos durante el embarazo y la lactancia sólo deberá contemplarse, si el beneficio esperado para la madre supera cualquier posible riesgo para el feto o niño. No se cuenta con suficiente experiencia en cuanto al uso de xinafoato de salmeterol y propionato de fluticasona durante el embarazo y la lactancia en seres humanos. Los estudios realizados en animales para evaluar la toxicidad en la reproducción, ya sea con el fármaco administrado como monoterapia o en combinación, revelaron los efectos fetales esperados a niveles excesivos de exposición sistémica a un glucocorticoesteroide y a un potente agonista de los receptores beta2 adrenérgicos. La vasta experiencia clínica que se tiene con estas clases de fármacos no ha revelado indicios de que los efectos estén relacionados con la administración de dosis terapéuticas. Ni el xinafoato de salmeterol ni el propionato de fluticasona, han exhibido potencial alguno de toxicidad genética. Después de administrar dosis terapéuticas inhaladas, las concentraciones plasmáticas de salmeterol y propionato de fluticasona son muy bajas, por lo cual es probable que sean correspondientemente bajas en la leche materna humana. Esta teoría se encuentra sustentada por estudios realizados en animales lactantes, en los cuales se detectaron bajas concentraciones medicamentosas en la leche. No se dispone de información relacionada con la leche materna humana.
Reacciones secundarias y adversas: A continuación se enlistan todas las reacciones adversas asociadas con los componentes individuales, xinafoato de salmeterol y propionato de fluticasona. No hay reacciones adversas adicionales atribuidas a la combinación cuando se compara con los perfiles de eventos adversos de los componentes individuales. Se enlistan más adelante los eventos adversos por órgano / sistema y frecuencia. Las frecuencias se definen como: muy comunes (≥ 1/10), comunes (≥1/100 a < 1/10), poco comunes (≥1/1,000 a < 1/100), raras (≥1/10,000 a < 1/1,000) y muy raras ( < 1/10,000). La mayor parte de las frecuencias se determinó del conjunto de los estudios clínicos, 23 en asma y 7 de los estudios en EPOC. No todos los eventos fueron reportados en los estudios clínicos. Para esos eventos, la frecuencia se calculó basándose en datos espontáneos. Datos de los Estudios Clínicos: Infecciones e infestaciones: Comunes: Candidiasis de boca y garganta, neumonía (en pacientes con EPOC). Raros: candidiasis esofágica. Trastornos del sistema inmunológico: Reacciones de hipersensibilidad: Poco comunes: Reacciones cutáneas de hipersensibilidad, disnea. Raros: Reacciones anafilácticas. Trastornos endocrinos: Posibles efectos sistémicos incluyen (véase Precauciones Generales): Poco comunes: Cataratas. Raros: Glaucoma. Trastornos del metabolismo y nutrición: Poco comunes: Hiperglucemia. Trastornos psiquiátricos: Poco comunes: Ansiedad, trastornos del sueño. Raros: Cambios de comportamiento, incluyendo hiperactividad e irritabilidad (predominantemente en niños). Trastornos de sistema nervioso: Muy comunes: Cefalea. (Véase Precauciones Generales). Poco comunes: Temblor. (Véase Precauciones Generales). Trastornos Cardiacos: Poco comunes: Palpitaciones (véase Precauciones Generales), taquicardia, fibrilación auricular. Raros: arritmias cardiacas incluyendo taquicardia supraventricular y extrasístoles. Trastornos respiratorios, torácicos y del mediastino: Comunes: Ronquera/disfonia. Poco comunes: Irritación de garganta. Trastornos de piel y tejido subcutáneos: Poco comunes: Contusiones. Trastornos músculo-esqueléticos y del tejido conectivo: Comunes: Calambres musculares, artralgia. Datos Post-comercialización: Trastornos del sistema inmunológico: Reacciones por hipersensibilidad como: Raras: Angioedema (principalmente facial y edema orofaríngeo) y broncoespasmo. Trastornos endocrinos: Posibles efectos sistémicos incluyen (véase Precauciones Generales): Raros: Síndrome de Cushing, manifestaciones Cushingoides, supresión suprarrenal, retardo en el crecimiento en niños y adolescentes, disminución en la densidad mineral ósea. Trastornos respiratorios, torácicos y del mediastino: Raros: Bronco-espasmo paradójico (véase Precauciones Generales).
Interacciones medicamentosas y de otro género: Deberá evitarse la administración concomitante de b-bloqueadores (selectivos o no selectivos), a menos, que haya motivos contundentes para su uso. En circunstancias normales, después de la administración por vía inhalada de Propionato de Fluticasona, se obtienen bajas concentraciones plasmáticas debido al extenso metabolismo de primer paso y a la alta eliminación sistémica mediada por la isoenzima 3A4 del citocromo P450 en el intestino y el hígado. En consecuencia, es poco probable que se presenten interacciones medicamentosas clínicamente significativas. Un estudio sobre interacciones medicamentosas, realizado en sujetos sanos, mostró que el Ritonavir (un inhibidor altamente potente de la isoenzima 3A4 del citocromo P450) tiene la capacidad de incrementar, de manera significativa, las concentraciones plasmáticas de Propionato de Fluticasona, dando como resultado reducciones muy marcadas en las concentraciones de cortisol sérico. Durante su uso posterior a la comercialización, ha habido comunicaciones de interacciones medicamnetosas clínicamente significativas en pacientes que reciben Propionato de Fluticasona y Ritonavir, mismas que originaron la aparición de efectos colaterales característicos de los corticoesteroides sistémicos, incluyendo síndrome de Cushing y deterioro suprerrenal. Por tanto, se debe evitar el uso concomitante del Propionato de Fluticasona con Ritonavir, a menos que el beneficio potencial para el paciente supere el riesgo de aquellos efectos colaterales característicos de los corticoesteroides sistémicos. Algunos estudios han demostrado que otros inhibidores de la isoenzima 3A4 del citocromo P450 producen aumentos insignificantes (Eritromicina) y leves (Ketoconazol) en la exposición sistémica al Propionato de Fluticasona, sin que se presenten reducciones notorias en las concentraciones séricas de cortisol. Sin embargo, se recomienda tener cuidado cuando se coadministren estos inhibidores con ya que existe un riesgo de que aumente el grado de exposición sistémica al propionato de fluticasona. La coadministración de ketoconazol y SEREVENT ® produce un aumento significativo en el grado de exposición plasmática al Salmeterol (1.4 veces Cmax y 15 veces el AUC), lo cual podría ocasionar una prolongación en el intervalo QTc. (Véase en Precauciones Generales).
Alteraciones en los resultados de pruebas de laboratorio: No se han reportado.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Datos Preclínicos de Seguridad: El xinafoato de salmeterol y el propionato de fluticasona se han evaluado extensamente en análisis de toxicidad realizada en animales. Se observaron toxicidades significativas sólo a dosis superiores a las recomendadas para uso en seres humanos, las cuales fueron las esperadas para un glucocorticoesteroide y un potente agonista de receptores beta2 adrenérgicos. En estudios realizados a largo plazo, el xinafoato de salmeterol indujo la aparición de tumores benignos del músculo liso en el mesovario de ratas y útero de ratonas. Los roedores son sensibles a la formación de estos tumores inducidos farmacológicamente. No se considera que el salmeterol represente algún riesgo oncogénico significativo para el hombre. La coadministración de salmeterol y propionato de fluticason