SIBELIUM*
MOKSHA8
Denominación genérica: Flunarizina
Forma farmacéutica y formulación: Cada tableta contiene: Clorhidrato de flunarizina equivalente a 5.0 mg de flunarizina, Excipiente cbp1 tableta.Clorhidrato de flunarizina equivalente a 10.0 mg de flunarizina, Excipiente cbp 1 tableta.
Indicaciones terapéuticas: Profilaxis de la migraña clásica (con aura) o común (sin aura). Tratamiento sintomático del vértigo vestibular debido al trastorno funcional del sistema vestibular diagnosticado.
Farmacocinética y farmacodinamia: Propiedades Farmacocinéticas. Absorción. El medicamento se absorbe fácilmente y alcanza concentraciones plasmáticas pico de 2 a 4 horas después de su administración. Consigue el estado estacionario después de 5 a 6 semanas. Flunarizina es bien absorbida ( > 80%) desde el tracto gastrointestinal y alcanza concentraciones plasmáticas pico de 2 a 4 horas después su administración oral. Bajo condiciones de acidez gástrica disminuida (pH gástrico alto), la biodisponibilidad podría ser moderadamente inferior. Las concentraciones plasmáticas de flunarizina alcanzan el estado estacionario después de, aproximadamente, 8 semanas de administración de múltiples dosis una vez al día y son casi 3 veces mayores que las observadas después de una sola dosis. Las concentraciones en estado estacionario son proporcionales sobre un rango de dosis de 5 mg a 30 mg. Distribución. Flunarizina se une en > 99% a las proteínas plasmáticas. Tiene un amplio volumen de distribución; aproximadamente 78 l/kg en sujetos sanos y 207 l/kg en pacientes epilépticos, lo cual indica una distribución amplia en el tejido extravascular. El medicamento cruza rápidamente la barrera hematoencefálica; las concentraciones cerebrales son, aproximadamente, 10 veces más altas que las plasmáticas. Metabolismo. Flunarizina es metabolizada en el hígado, por lo menos, en quince metabolitos. La vía metabólica principal es el CYP2D6. Eliminación. Flunarizina y sus metabolitos son eliminados por las heces a través de la bilis. Entre 24 y 48 horas después de su administración, aproximadamente de 3% a 5% de la dosis administrada se elimina en las heces sin cambios y como metabolitos. Menos de 1% se excreta sin cambios en la orina. Su vida media de eliminación terminal es altamente variable; oscila entre 5 y 15 horas en la mayoría de los individuos después de una dosis única. Algunos sujetos muestran concentraciones plasmáticas de flunarizina ( > 0.5 ng/ml) por un periodo prolongado (mayor a 30 días), posiblemente debido a la redistribución del medicamento en otros tejidos. Propiedades Farmacodinámicas. Grupo farmacoterapéutico: preparaciones antivertigo, código ATC: N07CA03. Efectos farmacodinámicos. Flunarizina es un antagonista selectivo de calcio. Este previene la sobrecarga celular de calcio mediante la reducción de la excesiva afluencia de calcio a través de la membrana. Flunarizina no tiene efectos sobre la contractilidad o conducción cardiaca.
Contraindicaciones: SIBELIUM® está contraindicado en pacientes con antecedentes de enfermedades depresivas o con síntomas previos de enfermedad de Parkinson u otros padecimientos extrapiramidales. (ver Precauciones Generales y Reacciones Adversas) Hipersensibilidad a flunarizina o a cualquiera de los excipientes.
Precauciones generales: El tratamiento con SIBELIUM® puede aumentar el riesgo de síntomas extrapiramidales, depresivos y manifestaciones de Parkinsonismo, particularmente, en personas predispuestas, tales como los pacientes de edad avanzada. Por lo tanto, deberá utilizarse con precaución en tales pacientes. En casos raros la fatiga puede aumentar durante la terapia con SIBELIUM®: en este evento, la terapia debe ser descontinuada. No deberá excederse la dosis recomendada. Los pacientes deberán ser observados en intervalos regulares, especialmente durante la terapia de mantenimiento, de manera que los síntomas extrapiramidales o depresivos puedan ser detectados en forma temprana y, en caso de presentarse, descontinuar el tratamiento. Si durante la terapia de mantenimiento los efectos terapéuticos disminuyen, el tratamiento también deberá descontinuarse (ver Dosis y vía de administración). Efectos sobre la habilidad de conducción y uso de maquinaria. Se puede manifestar somnolencia, especialmente al inicio del tratamiento, por lo que se deben tomar medidas de precaución durante la conducción y operación de maquinaria peligrosa.
Restricciones de uso durante el embarazo y la lactancia: Embarazo. La seguridad de SIBELIUM® durante el embarazo no ha sido establecida. Una evaluación de modelos animales indicó que no hay efectos dañinos directos o indirectos en la reproducción, desarrollo embrionario o fetal, en el curso de la gestación o el desarrollo peri y posnatal. Lactancia. Se debe evitar la lactancia al estar bajo tratamiento con SIBELIUM®. Estudios en perros lactantes han mostrado que flunarizina es excretada en la leche y la concentración en ésta es mayor que en el plasma. No existen datos de la excreción en la leche humana.
Reacciones secundarias y adversas: Se presentan las reacciones adversas durante toda esta sección, Son reacciones adversas y eventos adversos que se consideran razonablemente asociados con el uso de clorhidrato de flunarizina con base en la valoración completa de la información disponible sobre éstos. No es posible establecer de manera confiable la relación causal con clorhidrato de flunarizina en casos individuales. Sin embargo, debido a que los estudios clínicos se realizan en condiciones muy variables, no es posible comparar directamente las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento con las tasas de los estudios clínicos de otro, por lo que éstos no pueden reflejar las tasas observadas en la práctica clínica. Datos de estudios clínicos. Datos de estudios doble ciego controlados con placebo. Reacciones adversas reportadas con incidencia > 1%. La seguridad de SIBELIUM® (5 a 10 mg/día) fue evaluada en 500 sujetos (de los cuales, 247 fueron tratados con SIBELIUM® y 253 recibieron placebo) que participaron en dos estudios clínicos doble ciego paralelo, placebo-controlado, un estudio se enfocó en el tratamiento de la migraña y el otro en el tratamiento del vértigo. Las reacciones adversas reportadas por ≥ 1% de los sujetos tratados con SIBELIUM® en estos estudios se muestran en la Tabla 1.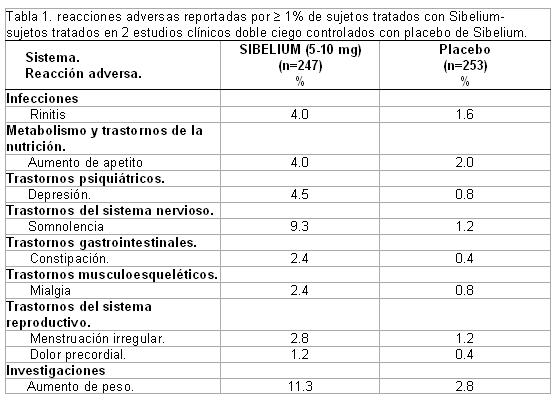
Datos comparativos-controlados activos - reacciones adversas reportadas con incidencia ≥1%. Dos estudios clínicos doble ciego controlados con comparador activo fueron seleccionados para determinar la incidencia de las reacciones adversas. En estos dos estudios, 476 sujetos fueron tratados con 10 mg/día de SIBELIUM®; uno en el tratamiento de migraña y el otro en el tratamiento de vértigo. Las reacciones adversas reportadas por ≥1% de sujetos tratado con SIBELIUM® observados en los estudios clínicos de comparador-activo controlado y no enlistados en la Tabla 1 se muestran en la Tabla 2.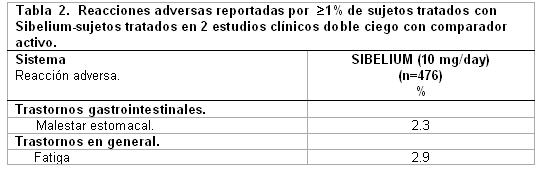
Datos de estudios controlados con placebo y comparador activo. Reacciones adversas reportadas con incidencia < 1%. La siguiente tabla muestra las reacciones adversas adicionales que ocurrieron en < 1% de sujetos tratados con SIBELIUM® en cualquiera de los dos estudios clínicos anteriores, las hojas de datos se enlistan en la Tabla 3.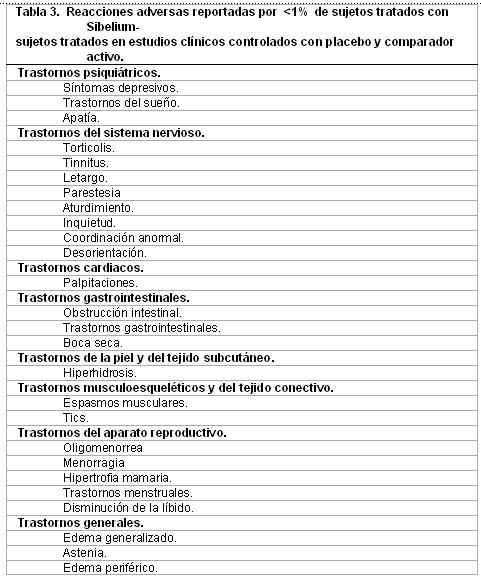
Datos postcomercialización. Los primeros eventos adversos identificados como reacciones adversas durante la experiencia postcomercialización con SIBELIUM® se incluyen en la Tabla 4. Las frecuencias son proporcionadas de acuerdo a la convención siguiente: Muy comunes ≥1/10, Comunes ≥1/100 a < 1/10, Poco comunes ≥1/1000 a < 1/100, Raras ≥1/10000 a < 1/1000, Muy raras < 1/10000, incluyen reportes aislados. En la tabla 4, las reacciones adversas son presentadas por categoría de frecuencia con base en las tasas de reportes espontáneos.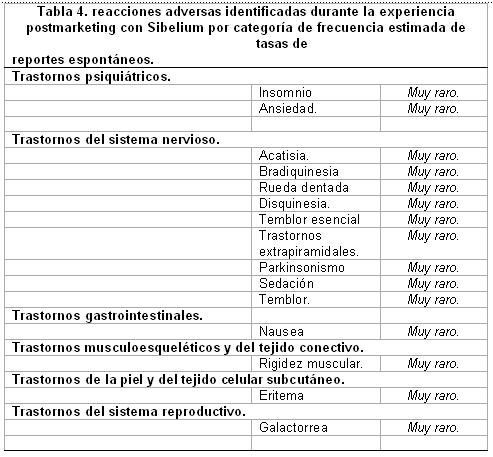
Interacciones medicamentosas y de otro género: Se puede presentar sedación excesiva cuando se administra SIBELIUM® simultáneamente con alcohol, hipnóticos o tranquilizantes. SIBELIUM® no está contraindicado en pacientes que utilicen betabloqueadores. La farmacocinética de flunarizina no se afecta por topiramato. Durante la coadministración de SIBELIUM® y topiramato 50 mg cada 12 horas, se observó un aumento de 16% de la concentración sérica de flunarizina en pacientes con migraña, comparado con 14% de aumento en pacientes que fueron tratados sólo con flunarizina. La farmacocinética de estado estable de topiramato no se afectó por la flunarizina. La administración crónica de flunarizina no afectó la biodisponibilidad de fenitoína, carbamazepina, valproato o fenobarbital. Las concentraciones plasmáticas de flunarizina fueron, generalmente, bajas en pacientes con epilepsia que tomaron estos medicamentos antiepilépticos en comparación con sujetos sanos que recibieron dosis similares. La unión de carbamazepina, valproato y fenitoína a proteínas plasmáticas no se afectó por la coadministración de flunarizina.
Alteraciones en los resultados de pruebas de laboratorio: Ninguna conocida.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Se observaron efectos preclínicos de índole del CNS (por ejemplo, sedación, salivación, ataxia) sólo en exposiciones consideradas suficientemente superiores a la exposición humana máxima, indicando poca relevancia para el uso clínico. Flunarizina ha sido probada en una serie extensiva de estudios de seguridad no clínicos incluyendo: dosis única de toxicidad después de administración oral (ratón, rata [adulto y joven], cobayo), intraperitoneal (ratón, rata), subcutáneamente (ratón, rata), intravenosamente (ratón, rata) e intra-arterial (rata): la dosis oral toxica repetida hasta 12 meses en perros y 18 meses en la rata e intravenosa toxica hasta 3 meses en el perro y 1 mes en la rata; estudios de reproducción vía oral que prueban la fertilidad y rendimiento general de reproducción en la rata; teratogenicidad y embriotoxicidad en ratas, y conejos y peri y post-natal en ratas. La mutagenicidad fue evaluada en una serie extensa de estudios, incluyendo: estudios de punto y/o de mutación genética in vitro en Salmonella typhimurium y en la prueba letal recesiva ligada al género en Drosophila melanogaster, estudios de aberración cromosómica in vitro en linfocitos humanos, prueba letal dominante y de micronúcleos in vivo en ratones. La carcinogenicidad ha sido evaluada en modelos de ratas y ratones después de mucho tiempo de administración oral. Los resultados de la toxicidad oral de dosis única (ratón -DL5' aproximadamente 960-1896 mg/kg; rata -DL50 aproximadamente 343-1935 mg/kg) indican un gran margen de seguridad cuando se compara a la dosis terapéutica humana máxima en una base de mg/kg (aproximadamente 0.2 mg/kg, basado en un paciente de 50 kg). Los resultados de la toxicidad oral a dosis repetida en ratas y perros muestran algunos efectos clínicos que podría ser relacionado a un efecto farmacológico exagerado, sin embargo, estos generalmente son vistos a dosis suficientemente cerca (aproximadamente 400 veces la dosis terapéutica humana máxima en una base de mg/kg) del rango de dosis fármaco-terapéutica e indica pequeña relevancia para uso clínico. En estudios de reproducción, no hubo efectos en la fertilidad y ni teratogenicidad. A dosis muy altas (aproximadamente 150-400 veces la dosis terapéutica humana máxima en una base de mg/kg) la fetotoxicidad fue secundaria a la toxicidad maternal. Flunarizina no es mutagénica y no se considera un carcinógeno primario. Sólo a niveles de dosis tóxicas en ratones (aproximadamente 50-100 veces la dosis terapéutica humana máxima en una base de mg/kg), se observaron ligeros efectos mediados por prolactina en el desarrollo de la glándula mamaria y tumorigénesis. En un modelo in vivo en cobayos anestesiados, flunarizina a dosis de 9.87 mg/kg intravenosamente (aproximadamente 50 veces la dosis terapéutica humana máxima, en una base de mg/kg) no tuvo efecto en el intervalo QTc ni en la morfología del ECG.
Dosis y vía de administración: Adultos y personas de edad avanzada (mayores de 18 años de edad). Profilaxis de migraña. Dosis inicial: El tratamiento se inicia con 10 mg/día (por las noches) en pacientes adultos de 18 a 64 años, y con 5 mg/día en pacientes de 65 años o más. Si durante el tratamiento se manifiestan síntomas de depresión, extrapiramidales u otros efectos adversos indeseables, la administración deberá descontinuarse (ver Precauciones generales). Si después de 2 meses no se observa alguna mejoría importante, se deberá considerar al paciente como no respondedor al tratamiento y deberá descontinuarse. Mantenimiento: Si el paciente está respondiendo satisfactoriamente y es necesario el tratamiento de mantenimiento, se deberá cambiar la dosificación de manera que éste reciba 5 días de tratamiento cada semana, la misma dosis diaria y 2 días sin medicamento cada semana. Aún en el caso de que el tratamiento profiláctico de mantenimiento sea exitoso y bien tolerado, éste deberá interrumpirse a los 6 meses y reiniciarse sólo si el paciente recae. Vértigo. Se deberá administrar la misma dosis que se utiliza para migraña, pero el tratamiento inicial para el control de los síntomas no deberá usarse más tiempo del necesario, lo cual generalmente toma menos de 2 meses. Si después de un mes de tratamiento para vértigo crónico, o después de 2 mes meses para vértigo paroxístico no se observa ninguna mejoría, se deberá considerar al paciente como no respondedor al tratamiento y la administración deberá descontinuarse.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Síntomas y signos. Con base en las propiedades farmacológicas del medicamento, podrían presentarse sedación y astenia. Han sido reportados pocos casos de sobredosis aguda (hasta 600 mg en una toma) y los síntomas observados fueron sedación, agitación y taquicardia. Tratamiento. No se conoce ningún antídoto específico. Se puede utilizar carbón activado si se considera apropiado.
Presentaciones: Caja con 20, 40 ó 60 tabletas de 5 mg o 10 mg en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a no más de 30° C. Protéjase de la luz.
Leyendas de protección: Su venta requiere receta médica. Literatura exclusiva para profesionales de la salud. No se deje al alcance de los niños. No se administre durante el embarazo y la lactancia. No se administre a menores de 18 años. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y atencionaclientes@its.jnj.com
Nombre y domicilio del laboratorio: Janssen-Cilag, S.A. de C.V. Carretera Federal México - Puebla km. 81.5, San Mateo Capultitlán, C.P. 74160, Huejotzingo, Puebla, México
Número de registro del medicamento: 444M96 SSA IV