SINCRONIUM®
FERRER
Denominación genérica: Acido acetilsalicílico; Simvastatina; Ramipril.
Forma famacéutica y formulación: Cápsulas. Cada cápsula contiene: Ácido acetilsalicílico 100 mg. Simvastatina 40 mg. Ramipril 2.5 mg. Excipiente, c.b.p. 1 cápsula. Cada cápsula contiene: Ácido acetilsalicílico 100 mg. Simvastatina 40 mg. Ramipril 5 mg. Excipiente, c.b.p. 1 cápsula. Cada cápsula contiene: Ácido acetilsalicílico 100 mg. Simvastatina 40 mg. Ramipril 10 mg. Excipiente, c.b.p. 1 cápsula.
Indicaciones terapéuticas: SINCRONIUM® está indicado para la prevención secundaria de accidentes cardiovasculares en aquellos pacientes en quienes esté indicado el uso combinado de simvastatina, ramipril y ácido acetilsalicílico.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: Grupo farmacoterapéutico: Inhibidores de la HMG-CoA reductasa, otras combinaciones. Ácido acetilsalicílico: El ácido acetilsalicílico inhibe de forma irreversible la agregación plaquetaria. Este efecto en las plaquetas se debe a la acetilación de la ciclooxigenasa, que inhibe de forma irreversible la síntesis del tromboxano A2 (una prostaglandina que favorece la agregación plaquetaria y la vasoconstricción) en las plaquetas. Este efecto es permanente y suele durar toda la vida de una plaqueta (aproximadamente 8 días). Paradójicamente, el ácido acetilsalicílico también inhibe la síntesis de la prostaciclina (una prostaglandina que inhibe la agregación plaquetaria, pero con efectos vasodilatadores) en las células endoteliales de los vasos sanguíneos; este efecto es reversible. En cuanto el ácido acetilsalicílico se ha eliminado de la sangre, las células endoteliales nucleadas vuelven a sintetizar prostaciclina. Como resultado, una única dosis diaria baja de ácido acetilsalicílico ( < 100 mg/día) inhibe el tromboxano A2 en las plaquetas sin alterar considerablemente la síntesis de prostaciclina. El ácido acetilsalicílico también pertenece al grupo de antiinflamatorio no esteroideos acídicos con propiedades analgésicas, antipiréticas y antiinflamatorias. Su mecanismo de acción consiste en la inhibición irreversible de las enzimas de la ciclooxigenasa implicadas en la síntesis de la prostaglandina. En dosis altas, el ácido acetilsalicílico se utiliza para el tratamiento del dolor leve a moderado, de la elevación de temperatura corporal y para el tratamiento de enfermedades inflamatorias agudas y crónicas, como la artritis reumatoide. Simvastatina: Después de la ingestión oral, la simvastatina se transporta activamente al interior de los hepatocitos por el transportador OATP1B1 y tiene una potente actividad inhibiendo la HMG-CoA reductasa (3 hidroxi-3-metilglutaril CoA reductasa). Esta enzima cataliza la conversión del HMG-CoA a mevalonato, un paso inicial y limitante de la biosíntesis del colesterol. Se ha demostrado que la simvastatina reduce las concentraciones normales y elevadas de C-LDL. La LDL se forma a partir de proteínas de densidad muy baja (VLDL) y se cataboliza predominantemente a través del receptor de LDL de gran afinidad. El mecanismo del efecto reductor LDL de la simvastatina puede implicar la reducción de la concentración del colesterol-VLDL (C-VLDL) y la inducción del receptor LDL, lo que produce una disminución de la producción y un aumento del catabolismo del C-LDL. La apolipoproteína B también disminuye sustancialmente durante el tratamiento con simvastatina. Además, la simvastatina aumenta moderadamente el C-HDL y reduce los TG plasmáticos. Como resultado de estos cambios, los cocientes entre C-total y C-HDL, y C-LDL y C-HDL se reducen. Alto riesgo de cardiopatía coronaria (CC) o cardiopatía coronaria existente: En el Heart Protection Study(HPS), los efectos del tratamiento con simvastatina fueron evaluados en 20,536 pacientes (edad entre 40-80 años), con o sin hiperlipidemia y con cardiopatía coronaria, otras enfermedades arteriales oclusivas o diabetes mellitus. En este estudio, 10,269 pacientes fueron tratados con simvastatina 40 mg/día y 10,267 pacientes fueron tratados con placebo durante un periodo medio de 5 años. Al principio, 6,793 pacientes (33%) tenían niveles de C-LDL por debajo de 116 mg/dl; 5,063 pacientes (25%) tenían niveles entre 116 y 135 mg/dl, y 8,680 pacientes (42%), tenían niveles superiores a 135 mg/dl. El tratamiento con simvastatina 40 mg/día comparado con un placebo redujo significativamente el riesgo de mortalidad por todas las causas (1,328 (12.9%) en pacientes tratados con simvastatina frente a 1,507 (14.7%) en pacientes tratados con placebo, p = 0,0003), debido a una reducción en el índice de muertes coronarias de 18% (587 [5.7%] frente a 707 [6.9%], p = 0,0005, reducción del riesgo absoluto de 1.2%). La reducción en las muertes no vasculares no alcanzó un significado estadístico. Simvastatina también disminuyó el riesgo de acontecimientos coronarios importantes (un criterio de valoración combinado que incluye infarto de miocardio no mortal o muerte por CC) en 27% (p < 0,0001). Simvastatina redujo la necesidad de tener que someterse a procedimientos de revascularización coronaria (incluyendo injerto de derivación de las arterias coronarias o angioplastia coronaria transluminal percutánea) y a procedimientos de revascularización periféricos y otros no coronarios en 30% (p < 0,0001) y 16% (p = 0,006), respectivamente. La simvastatina redujo el riesgo de accidente cerebrovascular en 25% (p < 0,0001), atribuible a una reducción en el accidente cerebrovascular isquémico de 30% (p < 0,0001). Ademas, dentro del subgrupo de pacientes con diabetes, simvastatima redujo el riesgo de desarrollar complicaciones macrovasculares, incluyendo procedimientos de revascularización periféricos (cirugía o angioplastia), amputaciones de miembros inferiores o úlceras en las piernas en 21% (p = 0,0293). Las reducciones proporcionales en el índice de acontecimientos fue similar en cada subgrupo de pacientes estudiados, incluyendo aquellos sin enfermedad coronaria, pero que tenían enfermedad cerebrovascular o arteriopatía periférica, hombres y mujeres, aquellos con edad inferior o superior a 70 años al entrar en el estudio, presencia o ausencia de hipertensión y principalmente aquellos con colesterol LDL por debajo de 3.0 mmol/L en la inclusión. En el scandinavian simvastatin survival study (4S), se valoró el efecto del tratamiento con simvastatina sobre la mortalidad total en 4,444 pacientes con CC y un nivel basal de colesterol total de 212-309 mg/dl (5.5-8.0 mmo/L). En este estudio multicéntrico, aleatorio, doble ciego y controlado con placebo, los pacientes con angina o infarto de miocardio (IM) previo fueron tratados con dieta, cuidados habituales y con 20-40 mg al día de simvastatina (n = 2,221) o placebo (n = 2,223) durante una duración media de 5.4 años. La simvastatina redujo el riesgo de muerte en 30% (reducción del riesgo absoluto de 3.3%). El riesgo de muerte por CC se redujo en 42% (reducción del riesgo absoluto de 3.5%). Simvastatina también redujo el riesgo de presentar episodios coronarios importantes (fallecimiento por CC más IM no fatal y silente diagnosticado hospitalariamente) en 34%. Además, simvastatina redujo significativamente el riesgo de acontecimientos cerebrovasculares mortales y no mortales (accidente cerebrovascular y ataques isquémicos transitorios) en 28%. No hubo diferencias estadísticamente significativas entre los grupos en la mortalidad no cardiovascular. Hipercolesterolemia primaria e hiperlipidemia combinada: En estudios que comparan la eficacia y la seguridad de simvastatina 10, 20, 40 y 80 mg al día en pacientes con hipercolesterolemia, las reducciones medias de C-LDL fueron de 30, 38, 41 y 47%, respectivamente. En estudios de pacientes con hiperlipidemia combinada (mixta) de simvastatina 40 y 80 mg, las reducciones medias en los triglicéridos fueron de 28 y 33% (placebo 2%), respectivamente, y los aumentos medios del C-HDL fueron de 13 y 16% (placebo 3%), respectivamente. Ramipril: El ramiprilato, el metabolito activo del profármaco ramipril, inhibe la enzima dipeptidilcarboxipeptidasa I (sinónimos: enzima conversora de la angiotensina, cininasa II). En plasma y tejidos, esta enzima cataliza la conversión de la angiotensina I en angiotensina II, la sustancia vasoconstrictora activa, así como la degradación de la bradiquinina, la sustancia vasodilatadora activa. La reducción de la formación de angiotensina II y la inhibición de la degradación de la bradiquinina resultan en vasodilatación. Dado que la angiotensina II también estimula la liberación de aldosterona, el ramiprilato reduce la secreción de aldosterona. La respuesta promedio a la monoterapia con un inhibidor de la ECA fue menor en pacientes hipertensos de raza negra (afro-caribeños, población hipertensa por lo general con renina baja) que en pacientes de raza no negra. Efectos farmacodinámicos: Propiedades antihipertensivas: La administración de ramipril provoca una marcada reducción de la resistencia arterial periférica. Por lo general, no se producen grandes cambios en flujo plasmático renal ni en la tasa de filtración glomerular. La administración de ramipril a pacientes con hipertensión resulta en la reducción de la presión arterial en supino y en bipedestación sin un aumento compensador de la frecuencia cardiaca. En la mayoría de pacientes, el comienzo del efecto antihipertensivo de una sola dosis se observa entre una y dos horas después de la administración oral. El efecto máximo de una sola dosis suele alcanzarse entre 3 y 6 horas después de la administración oral. El efecto antihipertensivo de una sola dosis dura por lo general 24 horas. El efecto antihipertensivo máximo del tratamiento continuado con ramipril se observa por lo general transcurridas entre tres y cuatro semanas. Se ha demostrado que el efecto antihipertensivo se mantiene en el tratamiento a largo plazo de dos años de duración. La suspensión abrupta del ramipril no conlleva un aumento de rebote rápido y excesivo de la presión arterial. Insuficiencia cardiaca: Además del tratamiento convencional con diuréticos y glucósidos cardiacos opcionales, se ha demostrado que ramipril es eficaz en pacientes con insuficiencia cardiaca de clases II-IV de la New York Heart Association. El fármaco tuvo efectos beneficiosos sobre la hemodinámica cardiaca (disminución de las presiones de llenado de ventrículos izquierdo y derecho, reducción de la resistencia vascular periférica total, aumento del gasto cardiaco y mejora del índice cardiaco). También redujo la activación neuroendocrina. Seguridad y eficacia clínica: Prevención cardiovascular/nefroprotección: Se ha llevado a cabo un ensayo preventivo controlado con placebo (el estudio HOPE), en el que se añadió ramipril al tratamiento estándar en más de 9,200 pacientes. En dicho ensayo se incluyeron pacientes con aumento del riesgo de enfermedad cardiovascular aterotrombótica (antecedentes de cardiopatía coronaria, ictus o vasculopatía periférica) o diabetes mellitus con al menos un factor de riesgo adicional microalbuminuria documentada, hipertensión, aumento del colesterol total, disminución de las lipoproteínas de alta densidad o hábito tabáquico). El ensayo mostró que ramipril, tanto en monoterapia como en combinación, redujo significativamente la incidencia de infarto de miocardio, muerte por causas cardiovasculares e ictus (eventos principales combinados).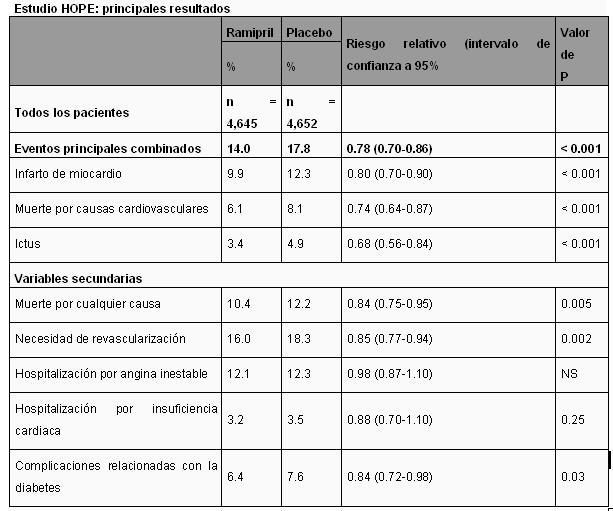
El ensayo MICRO-HOPE, un subestudio previamente definido dentro de HOPE, investigó el efecto de la adición de ramipril 10 mg al tratamiento médico que estaban recibiendo los pacientes en comparación con placebo en 3,577 pacientes de como mínimo ≥ 55 años (sin limite superior de edad), de los que la mayoría presentaba diabetes de tipo 2 (y como mínimo otro factor de riesgo CV), eran normotensos o hipertensos. El análisis principal mostró que 117 (6.5%) participantes con ramipril, y 149 (8.4%) con placebo desarrollaron una nefropatía franca, lo que equivale a una reducción del RR de 24% (IC a 95%, 3-40; p = 0.027). Por otro lado, el ensayo REIN, ensayo multicéntrico, aleatorizado, en doble ciego, de grupos paralelos y controlado con placebo, tuvo como objetivo evaluar el efecto del tratamiento con ramipril sobre el ritmo de reducción de la tasa de filtración glomerular (TFG) en 352 pacientes normotensos o hipertensos (de 18 a 70 años) con proteinuria leve (es decir, excreción urinaria media de proteínas > 1 y < 3 g/24 h) o grave (≥ 3 g/24 h) por nefropatía no diabética crónica. Ambas subpoblaciones se estratificaron prospectivamente. El análisis principal de los pacientes con la proteinuria de grado más grave (estrato prematuramente suspendido como consecuencia del beneficio observado en el grupo de ramipril) mostró que el ritmo medio de reducción de la TFG al mes era menor con ramipril que con placebo; -0.54 (0.66) vs. -0.88 (1.03) ml/min/mes, p = 0.038. Esto es, la diferencia entre los grupos fue de 0.34 [0.03-0.65] al mes y de unos 4 ml/min/año; 23.1% de los pacientes del grupo de ramipril alcanzó la variable combinada secundaria de una duplicación de la concentración sérica basal de creatinina y/o nefropatía en fase terminal (necesidad de diálisis o de trasplante renal) vs. 45.5% en el grupo de placebo (p = 0.02). Prevención secundaria tras infarto agudo de miocardio: El ensayo AIRE incluyó más de 2,000 pacientes con signos clínicos pasajeros/persistentes de insuficiencia cardiaca tras un infarto de miocardio documentado. El tratamiento con ramipril se inició de 3 a 10 días después del infarto de miocardio agudo. Tras un periodo de seguimiento medio de 15 meses, el ensayo mostró que la mortalidad en los pacientes tratados con ramipril era de 16.9%, mientras que en los pacientes tratados con placebo era de 22.6%, lo que significa una reducción de la mortalidad absoluta de 5.7% y una reducción del riesgo relativo de 27% (IC de 95%, [11-40%]). Propiedades farmacocinéticas: Ácido acetilsalicílico: El ácido acetilsalicílico se metaboliza en su principal metabolito activo, ácido salicílico, antes, durante y después de la absorción. Los metabolitos se eliminan básicamente por los riñones. Además del ácido salicílico, los metabolitos principales del ácido acetilsalicílico son el conjugado de glicina de ácido salicílico (ácido salicilúrico), el éter glucurónido y éster del ácido salicílico (glucurónido acetilsalicílico y salicilfenólico) y ácido gentísico formado por la oxidación del ácido salicílico y su conjugado de glicina. La absorción del ácido acetilsalicílico tras la administración oral es rápida y completa, en función de la formulación galénica. De hecho, la hidrólisis del residuo acetil del ácido acetilsalicílico tiene lugar, en cierto grado, durante el paso a través de la mucosa gastrointestinal. Las concentraciones plasmáticas máximas se alcanzan al cabo de 10-20 minutos (ácido acetilsalicílico) o al cabo de 0.3-2 horas, respectivamente (salicilato total). La cinética de eliminación del ácido acetilsalicílico depende en gran medida de la dosis, ya que la capacidad de metabolizar el ácido salicílico es limitada (la semivida de eliminación oscila entre 2 y 30 horas). La semivida de eliminación del ácido acetilsalicílico es de apenas unos minutos; la semivida de eliminación del ácido salicílico es de 2 horas después de la administración de una dosis de 0.5 g de ácido acetilsalicílico, 4 horas después de la administración de 1 g y aumenta a 20 horas tras una dosis única de 5 g. La unión a las proteínas plasmáticas en el ser humano depende de la concentración, se ha descrito que los valores oscilan entre 49% a más de 70% (ácido acetilsalicílico) y de 66 a 98% (ácido salicílico, respectivamente). El ácido salicílico se mide en disolución acuosa y líquido sinovial tras la administración de ácido acetilsalicílico. El ácido salicílico atraviesa la placenta y se excreta en la leche materna. Simvastatina: Simvastatina es una lactona inactiva que se hidroliza rápidamente in vivo al beta-hidroxiácido correspondiente, un inhibidor potente de la HMG-CoA reductasa. La hidrólisis tiene lugar principalmente en el hígado, el índice de hidrólisis en el plasma humano es muy bajo. Absorción: En el hombre, simvastatina se absorbe bien y sufre una extensa extracción hepática de primer paso. La extracción en el hígado depende del flujo sanguíneo hepático. El hígado es el lugar principal de acción de la forma activa. Se encontró que la biodisponibilidad del beta-hidroxiácido para la circulación sistémica después de una dosis oral de simvastatina era inferior a 5% de la dosis. La concentración plasmática máxima de los inhibidores activos se alcanzó 1-2 horas después de la administración de simvastatina. El consumo concomitante de alimentos no afecta a la absorción. La farmacocinética de las dosis únicas y múltiples de simvastatina mostró que no se producía acumulación del fármaco después de múltiples dosis. Distribución: La unión a proteínas de simvastatina y su metabolito activo es > 95%. Eliminación: Simvastatina es un sustrato de la CYP3A4 (véase Contraindicaciones). Los metabolitos principales de simvastatina presentes en el plasma humano son el beta-hidroxiácido y los cuatro metabolitos activos adicionales. Después de una dosis oral de simvastatina radiactiva en el hombre, 13% de la radiactividad se excretó en orina y 60% en las heces a lo largo de 96 horas. La cantidad recuperada en las heces representa equivalentes del medicamento absorbido excretado en la bilis, así como medicamento no adsorbido. Después de una inyección intravenosa del metabolito beta-hidroxiácido, su semivida es de un promedio de 1.9 horas. Un promedio de sólo 0.3% de la dosis I.V. se excretó en la orina como inhibidores. Ramipril: Absorción: Tras su administración oral, ramipril se absorbe rápidamente en el tracto gastrointestinal: las concentraciones plasmáticas máximas de ramipril se alcanzan en el plazo de una hora. De acuerdo a los datos de su recuperación urinaria, el grado de absorción es de mínimo 56% y no se ve influido de manera significativa por la presencia de alimentos en el tracto gastrointestinal. La biodisponibilidad del metabolito activo, ramiprilato, tras la administración oral de 2.5 y 5 mg de ramipril es de 45%. Las concentraciones plasmáticas máximas de ramiprilato, el único metabolito activo del ramipril, se alcanzan de 2 a 4 horas después de la toma del producto. Las concentraciones plasmáticas de ramiprilato en estado estacionario tras la administración una vez al día de las dosis habituales de ramipril, se alcanzan aproximadamente el cuarto día de tratamiento. Distribución: La unión a proteínas séricas de ramipril es de aproximadamente 73%, mientras que la de ramiprilato se encuentra en torno a 56%. Metabolismo: Ramipril se metaboliza casi completamente a ramiprilato, al éster dicetopiperazina, el ácido dicetopiperazina y los glucurónidos de ramipril y ramiprilato. Eliminación: La eliminación de los metabolitos es fundamentalmente renal. Las concentraciones plasmáticas de ramiprilato se reducen de manera polifásica. Como consecuencia de su unión potente y saturable a la ECA y de su lenta disociación de la enzima, el ramiprilato muestra una fase de eliminación terminal prolongada a concentraciones plasmáticas muy bajas. Tras la administración repetida de dosis únicas diarias de ramipril, la semivida efectiva de las concentraciones de ramiprilato fue de 13-17 horas con dosis de 5-10 mg y más prolongada con dosis más bajas, de 1.25-2.5 mg. Esta diferencia se debe a la capacidad saturable de la enzima que se une al ramiprilato. Una dosis oral única de ramipril no resultó en niveles detectables de ramipril y su metabolito en la leche materna. No obstante, se desconoce el efecto de dosis repetidas. Pacientes con insuficiencia renal (véase Dosis y vía de administración). La eliminación renal de ramiprilato es menor en los pacientes con insuficiencia renal, estando la eliminación de ramiprilato proporcionalmente relacionada con el aclaramiento de creatinina. Ello resulta en la elevación de las concentraciones de ramiprilato, que disminuyen más lentamente que en los sujetos con función renal normal. Pacientes con insuficiencia hepática (véase Dosis y vía de administración). En pacientes con insuficiencia hepática, la conversión metabólica de ramipril a ramiprilato fue más lenta debido a la disminución de la actividad de las esterasas hepáticas, por lo que los niveles plasmáticos de ramipril en estos pacientes estuvieron aumentados. No obstante, las concentraciones máximas de ramiprilato en estos pacientes no son diferentes de las observadas en sujetos con función hepática normal.
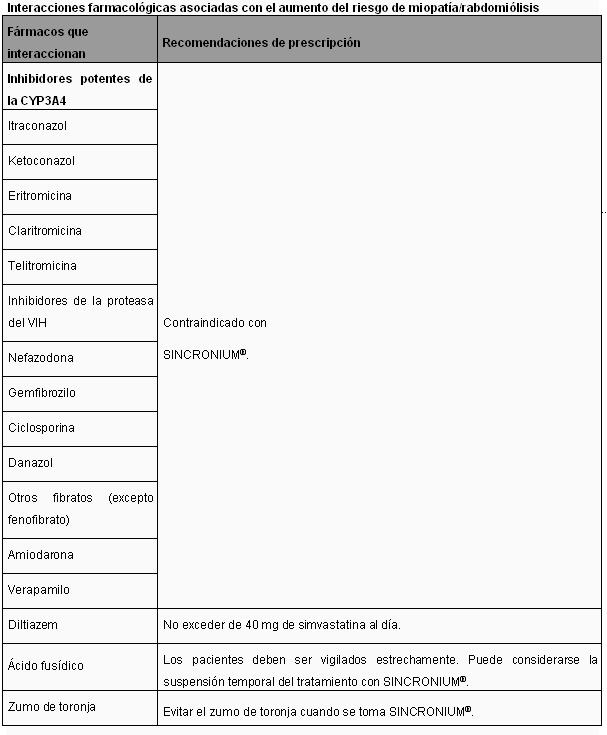
Contraindicaciones: Hipersensibilidad a los principios activos o a alguno de los excipientes, a otros salicilatos o a cualquier otro inhibidor de la ECA (enzima conversora de la angiotensina). En caso de antecedentes de crisis asmática u otra reacción alérgica al ácido salicílico y a otros analgésicos/antiinflamatorios no esteroideos. Úlceras intestinales y gástricas agudas (véase Precauciones generales). Hemofilia y otros trastornos de la coagulación. Insuficiencia hepática y renal grave (véase Dosis y vía de administración). Insuficiencia cardiaca grave. Tratamiento concomitante con metotrexato en dosis semanales iguales o superiores a 15 mg (véase Interacciones medicamentosas y de otro género). Hepatopatía activa o elevaciones persistentes e inexplicables de las transaminasas séricas (véase Precauciones generales). Embarazo y lactancia (véase Restricciones de uso durante el embarazo y la lactancia). Administración concomitante de inhibidores potentes de la CYP3A4 (por ejemplo, itraconazol, ketoconazol, inhibidores de la proteasa del VIH, eritromicina, claritromicina, telitromicina y nefazodona) (véase Interacciones medicamentosas y de otro género). Administración concomitante de gemfibrozilo, otros fibratos (excepto fenofibrato), ciclosporina, danazol, amiodarona y verapamilo. Antecedentes de angioedema (hereditario, idiopático o debido a un angioedema previo con inhibidores de la ECA o antagonistas de los receptores de la angiotensina II [ARAs II]). Tratamientos extracorpóreos que lleven al contacto de la sangre con superficies de carga negativa (véase Interacciones medicamentosas y de otro género). Estenosis bilateral importante de la arteria renal o estenosis de la arteria renal en caso de que funcione un solo riñón. Ramipril no debe emplearse en pacientes en situación de hipotensión o de inestabilidad hemodinámica. Niños y adolescentes menores de 18 años.
Precauciones generales: En los siguientes casos se requiere un control médico especialmente minucioso: Hipersensibilidad a otros analgésicos/antiinflamatorios/antirreumáticos o a otros alergenos (véase Contraindicaciones). Otras alergias conocidas (por ejemplo, reacciones cutáneas, prurito, urticaria), asma bronquial, rinitis alérgica, inflamación de las membranas mucosas nasales (hiperplasia adenoidea) y otras enfermedades respiratorias crónicas. Tratamiento concomitante con anticoagulantes. Pacientes con antecedentes de úlceras gástricas o intestinales o hemorragia gastrointestinal. Pacientes con disfunción hepática o renal (véase Dosis y vía de administración). Pacientes con riesgo de hiperuricemia. Las dosis bajas de ácido acetilsalicílico reducen la eliminación de ácido úrico, lo que puede desencadenar una crisis de gota. Miopatía/rabdomiólisis: Disminución de la función de proteinas transportadoras. Simvastatina, como otros inhibidores de la HMG-CoA reductasa, causa ocasionalmente miopatía, que se manifiesta como dolor, sensibilidad a la presión o debilidad musculares con valores de creatina quinasa (CK) superiores a diez veces el límite superior normal (LSN). La miopatía a veces toma la forma de rabdomiólisis con o sin insuficiencia renal aguda secundaria a mioglobinuria y muy raras veces se han producido muertes. El riesgo de miopatía aumenta con niveles altos en plasma de actividad inhibidora de la HMG-CoA reductasa. Pacientes con polimorfismo SLCO 1B1: La disminución de la actividad de proteínas transportadoras de aniones orgánicos (OATP) hepáticas puede aumentar la exposición sistémica a simvastatina y aumentar el riesgo de miopatía y rabdomiólisis. Esta reducción de la función hepática puede ocurrir como resultado de la inhibición por medicamentos (por ejemplo, cíclosporina) o en pacientes que son portadores del genotipo SLCO 1B1 alelo c.521T > C. Los pacientes portadores del alelpo c.521T>C, del gen SLCO que codifica una proteína OATP1B1 menos activa, tienen una mayor exposición sistémica a simvastatina relacionado con miopatía es de aproximadamente 1% en general, sin pruebas genéticas. En base a los resultaciós del ensayo SEARCH, los portadores del alelo homocigoto C (también denominado CC) tratados con 80 mg de simvastatina, tienen un riesgo del 15% de miopatia en un año, mientras que el riesgo en los portadores del alelo C heterocigoto (TC) es de 1.5%. El riesgo, correspondiente es de 0.3% en los pacientes que tienen el genotipo más común (TT) (véase Propiedades farmacocinéticas). Si se encuentra disponible, se debe considerar realizar un genotipo para conocer la presencia del alelo C, como parte de la evaluación del riesgo-beneficio antes de prescribir 80mg de simvastatina y evitar dosis altas para los pacientes que tengan el genotipo CC. Sin embargo, la ausencia de este gen en el genotipo no excluye que la miopatia pueda ocurrir. Como en otros inhibidores de la HMG-CoA reductasa, el riesgo de miopatía/rabdomiólisis está relacionado con la dosis. En la base de datos de un ensayo clínico en el que 41,050 pacientes fueron tratados con simvastatina, 24,747 (aproximadamente 60%) fueron tratados durante al menos 4 años, la incidencia de miopatía fue aproximadamente de 0.08% con 40 mg/día. En estos ensayos, los pacientes fueron cuidadosamente vigilados y se excluyeron algunos medicamentos que interaccionan. Determinación de la creatina quinasa: La creatina quinasa (CK) no debe ser determinada después de ejercicio extenuante o en presencia de cualquier causa alternativa aceptable de incremento de CK, ya que esto hace difícil la interpretación del valor. Si las concentraciones iniciales de CK están significativamente elevadas ( > 5 x LSN), se deben volver a determinar las concentraciones en un plazo de 5 a 7 días más tarde para confirmar los resultados. Mientras dure el tratamiento: Si aparece dolor muscular, debilidad o calambres musculares, mientras un paciente está recibiendo con una estatina, deben determinarse sus concentraciones de CK. Si se encuentra que estas concentraciones, en ausencia de ejercicio extenuante, están significativamente elevadas ( > 5 x LSN), se deberá interrumpir el tratamiento. Si los síntomas musculares son graves y producen malestar diario, incluso si las concentraciones de CK son de < 5 x LSN, debe considerarse la interrupción del tratamiento. Si se sospecha miopatía por cualquier otra razón, el tratamiento debe interrumpirse. Si los síntomas se resuelven y las concentraciones de CK se normalizan, se puede considerar la reintroducción de SINCRONIUM® en relación con un posible beneficio y con estrecha vigilancia. Medidas para reducir el riesgo de miopatía causada por interacciones con medicamentos: Véase Interacciones medicamentosas y de otro género. El uso concomitante de simvastatina con inhibidores potentes de la CYP3A4 (como itraconazol, ketoconazol, eritromicina, claritromicina, telitromicina, inhibidores de la proteasa del VIH, nefazodona), así como gemfibrozilo, ciclosporina, danazol y otros fibratos (excepto fenofibrato), amiodarona y verapamilo está contraindicado por el aumento significativo de miopatía y rabdomiólisis (véase Contraindicaciones). Se recomienda precaución al combinar simvastatina con diltiazem (véase Interacciones medicamentosas y de otro género). Debe evitarse tomar simvastatina con zumo de toronja. Se debe tener precaución cuando se receta fenofibrato o niacina (1 g/día) con simvastatina, ya que ambos fármacos pueden causar miopatía cuando se administran solos. Si la combinación se considera necesaria, los pacientes en tratamiento con ácido fusídico y simvastatina deben ser vigilados estrechamente (véase Interacciones medicamentosas y de otro género). Puede considerarse la suspensión temporal del tratamiento SINCRONIUM®. Enfermedad intersticial pulmonar: Se han notificado casos excepcionales de enfermedad intersticial pulmonar con algunas estatinas, especialmente en tratamientos de larga duración (véase Reacciones secundarias y adversas). Se presentan síntomas que pueden incluir disnea, tos no productiva y deterioro de la salud en general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad intersticial pulmonar, debe interrumpirse el tratamiento con SINCRONIUM®. Efectos hepáticos: En los estudios clínicos, se han producido aumentos persistentes (a > 3 x LSN) de las transaminasas séricas en unos pocos pacientes adultos que recibieron simvastatina. Cuando se interrumpió de modo temporal o definitivo la administración de simvastatina en estos pacientes, los niveles de las transaminasas normalmente descendían lentamente hasta los valores existentes antes del tratamiento. Se recomienda la realización de pruebas de función hepática antes de iniciar el tratamiento y después cuando esté clínicamente indicado. Debe prestarse una atención especial a los pacientes que desarrollan niveles séricos elevados de transaminasas, y en estos pacientes, deben repetirse las determinaciones rápidamente, y realizarse después con más frecuencia. Debe interrumpirse la administración de simvastatina cuando los niveles de transaminasas muestren indicios de progresión, especialmente si se elevan de forma persistente hasta 3 x LSN. Este fármaco debe utilizarse con precaución en los pacientes que consumen cantidades importantes de alcohol. Embarazo: SINCRONIUM® está contraindicado durante el embarazo. Cuando se diagnostique un embarazo, deberá interrumpirse inmediatamente el tratamiento y, si procede, se iniciará un tratamiento alternativo. Pacientes con un riesgo particular de hipotensión: Pacientes con alta activación del sistema- renina angiotensina-aldosterona: en pacientes con activación del sistema renina-angiotensina-aldosterona, es necesaria la supervisión médica con monitorización de la presión arterial para reducir el riesgo de un descenso pronunciado agudo de la presión arterial y un deterioro de la función renal debido a la inhibición de la ECA, por ejemplo en el caso de: pacientes con hipertensión severa. Pacientes con insuficiencia cardiaca congestiva descompensada. Pacientes con impedimento al flujo de llenado o vaciado ventricular izquierdo hemodinámicamente relevante (por ejemplo, estenosis aórtica o mitral). Pacientes con estenosis unilateral de la arteria renal con un segundo riñón funcionante. Pacientes con depleción de líquidos o sales, real o posible (incluidos los pacientes con diuréticos). Pacientes con cirrosis hepática y/o ascitis. Pacientes sometidos a cirugía mayor o durante la anestesia con agentes que producen hipotensión. Insuficiencia cardiaca transitoria o persistente posterior a IM. Pacientes con riesgo de isquemia cardiaca o cerebral en caso de hipotensión aguda. La fase inicial del tratamiento requiere una supervisión médica especial. Cirugía: El tratamiento con SINCRONIUM® se interrumpirá provisionalmente unos días antes de una intervención quirúrgica mayor programada y cuando sobrevenga cualquier afección quirúrgica o médica mayor. En el caso de intervenciones menores, como extracciones dentales, puede prolongar la hemorragia. Control de la función renal: Se recomienda un seguimiento minucioso de los pacientes con insuficiencia renal (ver Dosis y Vía de administración). Hay un riesgo de insuficiencia renal, en especial en pacientes con insuficiencia cardiaca congestiva o después de un trasplante renal. Angioedema: Se han notificado casos de angioedema en pacientes tratados con inhibidores de la ECA, incluido ramipril (véase Reacciones secundarias y adversas). En caso de angioedema, se debe suspender el tratamiento con SINCRONIUM®. Se debe instaurar rápidamente el tratamiento de urgencia. Debe mantenerse al paciente bajo observación durante 12-24 horas como mínimo, siendo dado de alta una vez resueltos completamente los síntomas. Se han notificado casos de angioedema intestinal en pacientes tratados con inhibidores de la ECA incluido ramipril (véase Reacciones secundarias y adversas). Estos pacientes aquejaron dolor abdominal (con o sin náuseas o vómitos). Reacciones anafilácticas durante la desensibilización: La probabilidad y gravedad de las reacciones anafilácticas y anafilactoides al veneno de insectos y otros alergenos son mayores bajo inhibición de la ECA. Antes de la desensibilización deberá considerarse la suspensión temporal de SINCRONIUM®. Hiperpotasemia: Se ha observado hiperpotasemia en algunos pacientes tratados con inhibidores de la ECA, incluido ramipril. Entre los pacientes con riesgo de hiperpotasemia se encuentran: pacientes con insuficiencia renal, pacientes mayores de 70 años, pacientes con diabetes mellitus no controlada o en tratamiento con sales de potasio, diuréticos que retienen potasio y otros principios activos que elevan el potásico plasmático, o en condiciones tales como deshidratación, descompensación cardiaca aguda o acidosis metabólica. Si se considera apropiado el uso concomitante de cualquiera de los agentes mencionados, se recomienda la determinación periódica del potasio sérico (véase Interacciones medicamentosas y de otro género). Neutropenia/agranulocitosis: Se han notificado casos raros de neutropenia/agranulocitosis, así como trombocitopenia y anemia, y también se ha notificado depresión de médula ósea. Se recomienda la determinación con frecuencia del recuento leucocitario, a fin de poder detectar una posible leucopenia. Se recomienda un control más repetido en la fase inicial del tratamiento y en los pacientes con afectación de la función renal, en aquellos otros con enfermedad del colágeno concomitante (por ejemplo, lupus eritematoso o esclerodermia) y en los tratados con medicamentos que pueden alterar el cuadro hemático (véase Interacciones medicamentosas y Reacciones secundarias y adversas). Diferencias étnicas: Los inhibidores de la ECA pueden provocar angioedema con mayor frecuencia en los pacientes de raza negra que en los de otras razas. Al igual que con otros inhibidores de la ECA, la eficacia hipotensora de ramipril puede ser menor en las personas de raza negra que las de otras razas, posiblemente por la mayor prevalencia de hipertensión con un bajo nivel de renina en la población negra hipertensa. Tos: Se ha notificado la aparición de tos con el uso de inhibidores de la ECA. La tos es característicamente no productiva. Persistente y se resuelve espontáneamente al interrumpir el tratamiento. La tos inducida por los inhibidores de la ECA debe considerarse como parte del diagnosticó diferencial de la tos. Excipientes: Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: SINCRONIUM® está contraindicado durante el embarazo (véase Contraindicaciones). La inhibición de la síntesis de las prostaglandinas puede tener efectos negativos sobre el embarazo y/o el desarrollo del embrión/feto. Los datos de estudios epidemiológicos demuestran un aumento del riesgo de muerte fetal, anomalías cardiacas y gastrosquisis tras la administración de inhibidores de la síntesis de las prostaglandinas al principio del embarazo. Presumiblemente, el riesgo aumenta en relación con la dosis y la duración del tratamiento. La experiencia previa con dosis diarias de 50-150 mg de ácido acetilsalicílico administrado a mujeres embarazadas en el segundo y tercer trimestre no ha demostrado inhibición del parto, aumento de la tendencia hemorrágica u oclusión prematura del ductus arteriosus. No se ha establecido la seguridad de simvastatina en mujeres embarazadas. No se han realizado estudios clínicos controlados con simvastatina en mujeres embarazadas. Se han recibido, aunque raramente, informes de anomalías congénitas después de la exposición intrauterina a inhibidores de la HMG CoA reductasa. Sin embargo, en un análisis de aproximadamente 200 embarazos seguidos prospectivamente expuestos durante el primer trimestre a simvastatina o a otro inhibidor de la HMG CoA reductasa estrechamente relacionado, la incidencia de anomalías congénitas fue comparable a la observada en la población general. Este número de embarazos fue estadísticamente suficiente para excluir un aumento de 2.5 veces o más en las anomalías congénitas en la incidencia general. Aunque no hay evidencia de que la incidencia de anomalías congénitas en la descendencia de pacientes que toman simvastatina u otro inhibidor de la HMG CoA reductasa estrechamente relacionado difiera de la observada en la población general, el tratamiento materno con simvastatina puede reducir los niveles fetales de mevalonato, que es un precursor de la biosíntesis del colesterol. La ateroesclerosis es un proceso crónico, y normalmente la interrupción de los fármacos hipolipemiantes durante el embarazo debe tener poco impacto sobre el riesgo a largo plazo asociado a la hipercolesterolemia primaria. La evidencia epidemiológica sobre el riesgo de teratogenicidad tras la exposición a los inhibidores de la ECA durante el primer trimestre del embarazo no ha sido concluyente; no obstante no se puede excluir un pequeño aumento del riesgo. Por ello, salvo que se considere esencial continuar el tratamiento con inhibidores de la ECA, las pacientes que estén planeando quedarse embarazadas deben cambiar a un tratamiento antihipertensivo alternativo que tenga un perfil de seguridad conocido para su uso durante el embarazo. Cuando se diagnostique un embarazo, debe interrumpirse inmediatamente el tratamiento con inhibidores de la ECA, y, si procede, iniciarse un tratamiento alternativo. Se sabe que la exposición a inhibidores de la ECA/antagonistas de los receptores de la angiotensina II (ARA II) durante el segundo y tercer trimestres del embarazo, induce fetotoxicidad humana (disminución de la función renal, oligohidramnios, retraso en la osificación del cráneo) y toxicidad neonatal (fallo renal, hipotensión, hiperpotasemia). (Véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Si se produce una exposición al inhibidor de la ECA a partir del segundo trimestre del embarazo, se recomienda realizar una prueba de ultrasonido de la función renal y del cráneo. Los lactantes cuyas madres han sido tratadas con los inhibidores de la ECA deben ser cuidadosamente monitorizados por si se produce hipotensión, oliguria e hiperpotasemia (véase Contraindicaciones y Precauciones generales). Por estas razones SINCRONIUM® no debe utilizarse en mujeres embarazadas, que quieran quedar embarazadas o que sospechen que lo están. El tratamiento con SINCRONIUM® debe interrumpirse durante el embarazo o hasta que se determine que la mujer no está embarazada. (Véase Contraindicaciones). Lactancia: Las pequeñas cantidades de ácido acetilsalicílico y sus metabolitos pasan a la leche materna Se desconoce si si