SURVTYK
PISA
Denominación genérica: Imatinib.
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Mesilato de Imatinib equivalente a 100 mg y 400 mg de Imatinib. Excipiente, c.b.p. 1 tableta.
Indicaciones terapéuticas: Está indicado en el tratamiento de: Pacientes adultos y pediátricos con leucemia mieloide crónica (LMC), recién diagnosticada, (para uso pediátrico véase dosis y vía de administración). Pacientes adultos y pediátricos con leucemia mieloide crónica (LMC), en crisis blástica fase acelerada, o en fase crónica tras el fracaso de un tratamiento con interferón alfa (para uso pediátrico véase dosis y vía de administración). Pacientes adultos con leucemia linfoblástica aguda cromosoma Filadelfia positivo (Ph+LLA) de diagnóstico reciente, en combinación con la quimioterapia. Pacientes adultos con leucemia linfoblástica aguda cromosoma Filadelfia positivo (Ph+LLA) recidivante o refractaria al tratamiento, en monoterapia. Pacientes adultos con síndromes mieloproliferativos o mielodisplasicos (SMP o SMD), asociado con reordenamiento del gen del receptor del factor de crecimiento derivado de plaquetas (PDGFR). Pacientes adultos con mastocitosis sistémica (SM) sin mutación D816V de c-Kit o con estado mutacional desconocido de c-Kit. Pacientes adultos con Síndrome Hipereosinofílico (HES) y/o leucemia eosinofílica crónica (CEL). Pacientes adultos con Dermatofibrosarcoma Protuberans (DFSP) irresecable, recurrente y/o metastásico.
Farmacocinética y farmacodinamia: El mesilato de Imatinib se absorbe bien después de su administración oral, alcanzando concentraciones plasmáticas máximas al cabo de 2 a 4 horas. La biodisponibilidad media es aproximadamente del 98%. Se ha calculado que el imatinib se une a las proteínas plasmáticas aproximadamente en un 95%. Las semividas de eliminación plasmáticas del imatinib y de su metabolito activo principal, el derivado piperazina N-desmetilado, son de unas 18 y 40 horas respectivamente. La principal enzima responsable de la metabolización del imatinib es la isoenzima CYP3A4 del citocromo P450; las isoenzimas CYP1A2, CYP2D6, CYP2C9 y CYP2C19 también participan en menor grado. Alrededor del 81% de una dosis se elimina en el plazo de 7 días en las heces (68%) y en la orina (13%). Se excreta mayoritariamente en forma de metabolitos, y sólo un 25% lo hace como fármaco inalterado. El mesilato de Imatinib es un inhibidor de la tirosincinasa que inhibe la tirosincinasa BCR-ABL creada por una alteración del cromosoma Filadelfia en casos de leucemia mieloide crónica. El imatinib induce apoptosis y la proliferación de líneas celulares positivas bcr-abl y células leucémicas nuevas de la LMC cromosoma Filadelfia positivo. Imatinib fue evaluado en ensayos clínicos en fase I en los que participaron pacientes con leucemia mieloide crónica (LMC) en fase crónica que eran resistentes a IFN-alfa (Interferón alfa) o que no toleraban éste fármaco. En un estudio en fase I se obtuvieron efectos terapéuticos significativos con efectos con efectos adversos mínimos con dosis de 300 mg diarios y superiores. Según estos datos, se inició un ensayo clínico en fase II en el que participaron 454 pacientes con enfermedad en fase crónica confirmada y que presentaban fracaso terapéutico o intolerancia a interferon alfa. Estos pacientes fueron tratados con una dosis de imatinib de 400 mg diarios por vía oral. En éste estudio participaron pacientes con hasta un 15% de blastos y un 15% de basófilos en la médula ósea o la sangre periférica. La mediana de la duración de la enfermedad era de 34 meses y la del tratamiento previo con interferon alfa de 14 meses. Tras una mediana de seguimiento de 29 meses, se alcanzó una respuesta hematológica completa (RHC) en el 96% de los pacientes, con una mediana hasta la RHC inferior a 1 mes. Imatinib indujo la aparición de respuestas citogenéticas mayores en el 64% de los pacientes con una tasa de respuesta citogenética completa del 48%. La estimación de la supervivencia sin progresión de la enfermedad a los 24 meses fue del 87% (tabla 1).
En este estudio en fase II, las respuestas citogenéticas se han mantenido hasta el momento y se han correlacionado con un incremento de la supervivencia sin progresión de la enfermedad y la supervivencia global. Así, una vez que se alcanza una respuesta citogenética mayor, se calcula que el 91% de estos pacientes no presenta progresión de la enfermedad al cabo de 24 meses. La obtención de una respuesta citogenética mayor a los 3, 6 ó 12 meses se ha asociado a una mejora estadísticamente significativa de la supervivencia global. En un estudio en fase III con asignación aleatoria realizado entre junio de 2000 y enero de 2001 participaron 1106 pacientes con LMC recién diagnosticada en los que se comparó la administración de imatinib (400 mg al día) con la de interferon alfa más citarabina. En cada grupo de tratamiento participaron 553 pacientes. Las características iníciales estaban bien equilibradas entre ambos grupos con respecto a todas las características evaluadas, incluidas la edad, el recuento leucocítico, las puntuaciones de Sokal y Euro y el tiempo trascurrido desde el diagnóstico. Después de una mediana de seguimiento de 19 meses, los pacientes asignados aleatoriamente al grupo de imatinib mostraron resultados significativamente mejores que los tratados con la combinación de interferon alfa y citarabina respecto a todos los parámetros contemplados (tabla 2), incluidas las tasas de respuesta hematológica completa RHC (97% frente a 56%; P < 0,001), las respuestas citogenéticas mayor y completa (85% y 74% frente al 22% y 8%; P < 0,001), la necesidad de interrupción del tratamiento asignado debido a falta de tolerancia (3% frente al 31%) y la progresión a un cuadro de fase acelerada o una crisis blástica (3% frente al 8%; P < 0,001). Aunque se alcanzó una respuesta citogenética completa en el 74% de los asignados aleatoriamente al grupo de imatinib, la mayor parte de estos pacientes presentaba leucemia detectable según el análisis de transcritos de BCR-ABL en la RT-PCR.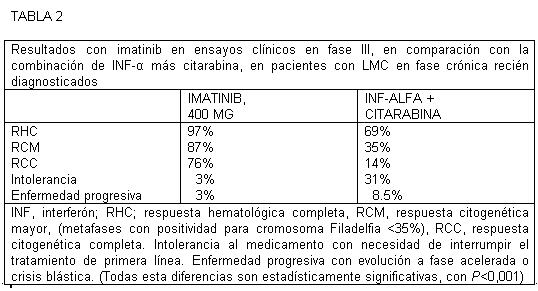
Contraindicaciones: Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Precauciones generales: Se debe tomar con alimentos y con bastante agua, en aquellos pacientes con dificultad para deglutir los comprimidos, se recomienda disolverlos en un vaso de agua o jugo de manzana (por ejemplo en 50 ml para un comprimido de 100 mg y 200 ml para un comprimido de 400 mg) Agitar con una cuchara hasta que se desintegre y administrar inmediatamente. No triturar los comprimidos y evitar el contacto con los comprimidos triturados, lavar muy bien si se produce un contacto directo de los comprimidos triturados con la piel o las mucosas, debe evitarse la ingestión de imatinib con jugo de uva. Debe evitarse el uso concomitante de fármacos inductores potentes del CYP3A4 (por ejemplo dexametasona, fenitoína, carbamazepina, rifampicina y fenobarbital) en caso de ser necesario el uso concomitante de estos fármacos se deberá reducir la dosis y monitorear al paciente. Se han registrado casos de severa disfunción ventricular izquierda e insuficiencia cardiaca congestiva, éste riesgo se incrementa en los pacientes con antecedentes de enfermedad cardiaca y pacientes ancianos, deben monitorearse de cerca los pacientes con enfermedad cardiaca o con factores de riesgo para la insuficiencia cardiaca o pacientes con antecedentes de insuficiencia renal. Se han reportado citopenias grado 3 y 4 (por ejemplo, anemia, neutropenia y trombocitopenia) por lo que se recomienda monitorear al paciente. También se han reportado reacciones dermatológicas como eritema multiforme y síndrome de Stevens-Johnson. Se han registrado casos de severa retención de líquidos y edema, el riesgo se incrementa en pacientes mayores de 65 años y con dosis altas, por lo que se recomienda el monitoreo de éstos pacientes. Se recomienda monitorear a los pacientes de edad avanzada debido a que tienen un incremento en el riesgo de desarrollar retención de líquidos, edema, severa disfunción ventricular izquierda e insuficiencia cardiaca congestiva. Se recomienda reducir la dosis en caso de presentarse disfunción hepática. Con el tratamiento a corto y a largo plazo han sido reportados casos de hepatotoxicidad, lesión hepática severa e insuficiencia hepática fatal que requieren de trasplante por lo que se recomienda un monitoreo continuo. Se ha reportado al inicio de la terapia síndrome hipereosinofílico con infiltración del células al miocardio, choque cardiogénico y disfunción ventricular izquierda, el riesgo se incrementa en pacientes con enfermedad mieloproliferativa y mielodisplásica y mastocitosis sistémica, se recomienda tratamiento profiláctico y monitoreo; se puede requerir la interrupción del tratamiento. Se recomienda monitorear a los pacientes pediátricos y pre-adolescentes debido a que se ha reportado retraso en el crecimiento. Debe prevenirse el embarazo con un método anticonceptivo efectivo durante el tratamiento ya que puede causar daño fetal. Se recomienda reducir la dosis en caso de presentarse disfunción renal. Se recomienda monitorear a los pacientes con tiroidectomía y medicados concomitantemente con levotiroxina ya que se ha reportado que clínicamente desarrollan hipotiroidismo. Deben ser monitoreados los pacientes que previo al tratamiento presentan tumores altamente proliferativos o con gran carga tumoral ya que se ha reportado síndrome de lisis tumoral con desenlace fatal. Manejo de maquinaria de precisión y vehículos: Puede ocurrir somnolencia, mareos y visión borrosa durante el tratamiento con imatinib que afectan la capacidad de conducir vehículos de motor, así pues debe aconsejarse cautela a la hora de conducir vehículos u operar máquinas de precisión.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: La FDA (Food and Drug Administration por sus siglas en ingles) en Estados Unidos lo ha clasificado en categoría D para todos los trimestres. Hay evidencia positiva de riesgo fetal en humanos sin embargo los beneficios de su uso en mujeres embarazadas puede ser aceptable a pesar del riesgo (por ejemplo si el fármaco es necesario en una situación que amenace la vida o en una grave enfermedad para la que medicamentos más seguros no se pueden utilizar o son ineficaces). Mujeres en edad de procrear: Se recomienda que debe prevenirse el embarazo con un método anticonceptivo eficaz durante el tratamiento ya que puede causar daño fetal. Lactancia: Los datos disponibles y / o el consenso de expertos no es concluyente o es insuficiente para determinar el riesgo infantil cuando se utiliza durante la lactancia. Se deben evaluar los beneficios potenciales del tratamiento contra los riesgos potenciales antes de prescribir este medicamento durante la lactancia. Imatinib y su metabolito activo se excretan en la leche humana, y los datos indican una relación de leche/plasma de alrededor de 0.5 y 0.9, respectivamente. Con base en el peso corporal, un bebé alimentado con leche materna podría recibir hasta un 10% de la dosis terapéutica materna cuando se combinan las concentraciones de ambos componentes. Se debe evaluar y decidir si se suspende la lactancia o suspender el fármaco, teniendo en cuenta la importancia del medicamento para la madre.
Reacciones secundarias y adversas: Entre los efectos adversos más habituales del mesilato de imatinib se incluyen trastornos gastrointestinales, edema superficial, mialgia, calambres musculares, exantema y cefalalgia. La depresión de la médula ósea, expresada en forma de neutropenia, trombocitopenia, o anemia, ocurre con más frecuencia en pacientes leucémicos, y puede ir asociada a la enfermedad subyacente. Puede aparecer retención grave de líquidos, que puede provocar un derrame pleural y pericárdico, edema pulmonar y ascitis, puede ser necesario interrumpir el tratamiento si se produce un aumento rápido e inesperado del peso corporal. Los pacientes de edad avanzada y los que tienen antecedentes de enfermedad cardíaca pueden presentar mayor riesgo. Raramente se ha descrito necrosis aséptica del hueso, principalmente en la cabeza del fémur. A continuación se encuentra un listado de las reacciones adversas reportadas, clasificadas por sistemas orgánicos: Efectos Cardiovasculares: Taponamiento cardiaco, choque carcinogénico, dolor precordial, insuficiencia cardiaca congestiva, edema, hipotensión, palpitaciones, derrame pericárdico, pericarditis, edema periférico, taquicardia. Efectos dermatológicos: Alopecia, erupción ampollosa, dermatitis, trastornos de la piel como rash cutáneo, piel seca, edema de cara, eritema multiforme, eritrodermia, repigmentación del cabello, carcinoma intraepitelial celular escamoso, dermatitis liquenoide, sudores nocturnos, fotosensibilidad, prurito, pseudoporfiria, psoriasis, rash exfoliativo, hipopigmentación de la piel, síndrome de Stevens-Johnson, necrólisis epidérmica toxica. Efectos endocrino-metabólicos: Retraso en el crecimiento corporal, hipoalbuminemia, hipoglicemia, hipocalemia, hipofosfatemia, hipoproteinemia, niveles séricos elevados de deshidrogenasa láctica, pérdida o incremento de peso. Efectos gastrointestinales: Dolor abdominal, obstrucción intestinal, constipación, diarrea, flatulencia, reflujo gastro-esofágico, perforación gastrointestinal, indigestión, pérdida del apetito, náusea, pancreatitis, estomatitis, distensión abdominal, alteración del sentido del gusto, ulceras en mucosa oral, vomito. Efectos hematológicos: Anemia, necrosis de la médula ósea, disminución de las cifras de hemoglobina, plaquetas y leucocitos, neutropenia febril, hemorragia, neutropenia, pancitopenia, trombocitopenia, trastornos tromboembolicos. Efectos hepáticos: Elevación de las enzimas hepáticas, ascitis, necrosis hepática, hepatotoxicidad, insuficiencia hepática, elevación de la fosfatasa alcalina sérica, elevación de bilirrubinas séricas. Efectos inmunológicos: Enfermedades infecciosas. Efectos musculoesqueléticos: Artralgias, calambres, derrame articular, hipofosfatemia, dolor musculoesquelético, mialgias, dolor en extremidades, espasmos. Efectos neurológicos: Astenia, edema cerebral, confusión, mareos, cefalea, insomnio, parestesia, incremento de la presión intracraneal, convulsiones, hematoma subdural. Efectos oftalmológicos: Visión borrosa, conjuntivitis, excesiva producción de lágrimas, glaucoma, papiledema, edema periorbital, edema de la retina, alteraciones visuales, hemorragia vítrea. Efectos óticos: Pérdida de la audición, pérdida auditiva neurosensorial. Efectos psiquiátricos: Depresión. Efectos renales: Insuficiencia renal aguda, incremento de la creatinina sérica. Efectos respiratorios: Insuficiencia respiratoria aguda, tos, disnea, hipoxia, nasofaringitis, dolor faringolaríngeo, faringitis, derrame pleural, neumonía, neumonitis, edema pulmonar, sinusitis, infecciones de aparato respiratorio alto. Otros: Fatiga, fiebre, influenza, carcinoma de células escamosas intraepiteliales, dolor, escalofríos, ruptura del bazo, enfermedades neoplásicas malignas secundarias, síndrome de lisis tumoral. Los pacientes con malignidades en estado avanzado pueden presentar numerosos trastornos que confunden u obstaculizan la determinación de la causalidad de los eventos adversos debido a la multiplicidad de síntomas relacionados con la enfermedad subyacente, la progresión de esta enfermedad y la coadministración de muchos medicamentos.
Interacciones medicamentosas y de otro género: El mesilato de imatinib es metabolizado por la isoenzima CYP3A4 del citocromo P450, y los fármacos que inhiben este sistema enzimático, como los antimicóticos azólicos y los antibacterianos macrólidos, pueden incrementar las concentraciones sanguíneas de imatinib. De la misma manera, los inductores de este sistema enzimático (como la carbamazepina, la dexametasona, el fenobarbital, la fenitoína y la rifampicina) pueden disminuir las concentraciones sanguíneas de imatinib. Estudios in vitro han demostrado que el propio imatinib inhibe las isoenzimas CYP3A4, CYP2C9 y CYP2D6 del citocromo P450, y que puede aumentar las concentraciones sanguíneas del fármaco que sean sustrato de dichas enzimas. El uso concomitante de imatinib con amiodarona provoca un incremento de las concentraciones plasmáticas de ambos fármacos. Se incrementa el riesgo de toxicidad con imatinib cuando se usa concomitantemente con amlodipino. Se presenta un incremento de los niveles plasmáticos de imatinib cuando se administra concomitantemente con aprepitant, claritromicina, dexametasona, eritromicina, itraconazol, ketoconazol, voriconazol y con jugo de uva. Disminuyen de los niveles plasmáticos de imatinib cuando se administra concomitantemente con carbamazepina, fenobarbital, fenitoína, rifabutina y rifampicina. Se incrementan las concentraciones plasmáticas y los efectos adversos de los siguientes fármacos: clozapina y fentanilo cuando se administran concomitantemente con imatinib. Se incrementan los niveles plasmáticos de los fármacos siguientes: alfentanilo, ciclosporina, dihidroergotamina, ergotamina, metoprolol, nimodipino, quinidina, simvastatina, sirolimus, tacrolimus, tamsulosina, cuando se administran concomitantemente con imatinib. Se incrementa los niveles plasmáticos de domperidona y el riesgo de prolongación del intervalo QT cuando se administra concomitante con imatinib. Se incrementa el riesgo de hepatotoxicidad el uso concomitante de imatinib con ginseng. Se reduce la eficacia de la levotiroxina y el empeoramiento del hipotiroidismo cuando se administra levotiroxina con imatinib. El uso concomitante de imatinib con warfarina incrementa el riesgo de sangrado.
Alteraciones en los resultados de pruebas de laboratorio: Hematológicas: Se debe realizar biometría hemática completa semanalmente durante el primer mes, cada dos semanas en el segundo mes y posteriormente de forma periódica, por el riesgo de depresión de la médula ósea, expresada en forma de neutropenia, trombocitopenia o anemia que ocurre más frecuentemente en pacientes leucémicos, y puede ir asociada a la enfermedad subyacente. Bioquímicas: Monitorear las pruebas de función hepáticas (transaminasas, bilirrubina, fosfatasa alcalina) al inicio del tratamiento y después mensualmente. Monitorear los niveles de hormona estimulante de tiroides (TSH) en pacientes tiroidectomizados que reciben terapia de reemplazo con levotiroxina durante el tratamiento con imatinib. Imatinib y sus metabolitos no se excretan por vía renal en un grado significativo. Se sabe que la depuración de creatinina se reduce con la edad y que ésta última no afecta de forma considerable la cinética de imatinib. En los pacientes con insuficiencia renal, la exposición plasmática al imatinib parece ser mayor que en los que tienen una función renal normal, probablemente porque tales pacientes presentan concentraciones plasmáticas elevadas de alfa glucoproteína ácida, proteína a la que se une el imatinib, en estos pacientes. Según la clasificación basada en la depuración de creatinina entre los pacientes con insuficiencia renal leve (depuración de creatinina de 40-59 ml/min) e insuficiencia renal grave (depuración de creatinina menor de 20 ml/min), no existe una correlación entre la exposición al imatinib y el grado de insuficiencia renal. Sin embargo no puede darse una recomendación específica respecto al ajuste de dosis, dado que no se han realizado estudios clínicos en pacientes con deterioro en la función renal.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Estudios realizados en animales con imatinib han mostrado evidencia de teratogenicidad potencial, la exposición sistémica de imatinib en ratas en dosis aproximadamente iguales a la dosis terapéutica máxima en humanos de 800 mg/día mostro evidencia de teratogenicidad. En la exposición sistémica de imatinib en dosis superiores a la dosis terapéutica máxima en humanos, todos los animales experimentaron una pérdida fetal. Las dosis más bajas que o iguales a un tercio de la dosis máxima en humanos no mostraron evidencia de pérdida fetal. No existen estudios adecuados y bien controlados del uso de imatinib en mujeres embarazadas, un estudio clínico reportó 16 abortos espontáneos o terapéuticos resultantes de 26 embarazos que se produjeron mientras la madre estaba tomando imatinib, en dos reportes de caso se trataba de mujeres con antecedentes de leucemia mieloide crónica que fueron tratadas con imatinib durante el periodo gestacional medio o tardío, sus bebés nacieron sanos y a término con poco o nada de imatinib detectado en la sangre del cordón umbilical. En otro reporte de caso una paciente femenina fue tratada con imatinib durante los días 8 al 33 del período gestacional, el bebé nació sano y a término, se diagnosticó una estenosis pilórica en el niño que requirió de cirugía a las 8 semanas y tuvo una recuperación total sin secuelas; son varios reportes de casos con resultados satisfactorios en las mujeres embarazadas expuestas a imatinib aunque también se han reportado abortos espontáneos y anomalías congénitas. Por lo tanto se debe recomendar a las mujeres en edad fértil que deben evitar el embarazo mientras están bajo tratamiento con imatinib, las pacientes sexualmente activas deben utilizar métodos anticonceptivos adecuados y seguros. Si una paciente queda embarazada mientras está tomando imatinib se le debe informar sobre el riesgo potencial para el feto.
Dosis y vía de administración: La dosis prescrita deberá ser administrada por vía oral. El tratamiento debe ser iniciado por un médico experimentado en el tratamiento de pacientes con procesos malignos hematológicos y sarcomas malignos, según el caso. Las dosis de 400 mg ó 600 mg deben administrarse una vez al día, mientras que una dosis diaria de 800 mg debe administrarse en dosis de 400 mg dos veces al día, por la mañana y por la noche. Se debe tomar con alimentos y con bastante agua, en aquellos pacientes con dificultad para deglutir los comprimidos, se recomienda disolverlos en un vaso de agua o jugo de manzana (por ejemplo en 50 ml para un comprimido de 100 mg y 200 ml para un comprimido de 400 mg) Agitar con una cuchara hasta que se desintegre y administrar inmediatamente. No triturar los comprimidos y evitar el contacto con los comprimidos triturados, lavar muy bien si se produce un contacto directo de los comprimidos triturados con la piel o las mucosas, debe evitarse la ingestión de imatinib con jugo de uva. Dosis en Leucemia Mieloide Crónica LMC: La dosis recomendada de imatinib es de 400 mg/día para pacientes con LMC en fase crónica y 600 mg/día para pacientes en fase acelerada o crisis blásticas. El tratamiento debe continuarse mientras el paciente siga beneficiándose con el mismo. El aumento de la dosis de 400 mg a 600 mg ó 800 mg en pacientes con enfermedad en fase crónica o de 600 mg hasta un máximo de 800 mg diariamente (administrados en dosis de 400 mg dos veces al día), en pacientes en fase acelerada o crisis blástica, puede considerarse en ausencia de reacciones medicamentosas adversas severas y neutropenia no relacionada con la leucemia severa o trombocitopenia en las siguientes circunstancias: progresión de la enfermedad (en cualquier momento); fracaso para alcanzar una respuesta hematológica satisfactoria después de por lo menos 3 meses de tratamiento; imposibilidad para obtener una respuesta citogenética después de 12 meses de tratamiento, o pérdida de una respuesta hematológica y/o citogenética previamente alcanzada. La dosis en los niños debe adaptarse a la superficie corporal (mg/m2), en los niños con LMC en fase crónica y avanzada se recomienda administrar dosis diarias de 340 mg/m2 (no exceder la dosis total de 600 mg/día). Se puede administrar una sola dosis diaria o se puede dividir la dosis diaria en dos tomas, una matutina y la otra vespertina. La dosis recomendada actual se basa en un pequeño número de pacientes pediátricos. No se tienen antecedentes del uso de imatinib en niños menores de 2 años de edad. Dosis en leucemia linfoblástica aguda cromosoma Filadelfia positivo (Ph+LLA): La dosis recomendada de imatinib es de 600 mg diarios. Dosis en síndrome mielodisplasico/síndrome mieloproliferativo: La dosis recomendada de imatinib es de 400 mg una vez al día para pacientes con SMD/SMP. Dosis en mastocitosis sistémica (SM); La dosis recomendada de imatinib es de 400 mg diarios para pacientes con mastocitosis sistémica sin la mutación D816V c-Kit. Si no se conoce o no está disponible el estado mutacional de c-Kit, se puede considerar el tratamiento con imatinib a dosis de 400 mg diarios que no respondan satisfactoriamente a otros tratamientos. Para pacientes con mastocitosis sistémica asociada a eosinofilia, una enfermedad hematológica relacionada con la cinasa de fusión FIP1L1-PDGFR alfa, se recomienda una dosis inicial de imatinib de 100 mg a 400 mg para estos pacientes en ausencia de reacciones adversas al fármaco si las valoraciones demuestran una respuesta insuficiente al tratamiento. Dosis en leucemia eosinofílica crónica (CEL) y en Síndrome Hipereosinofílico (HES): La dosis recomendada de imatinib es de 400 mg una vez al día para pacientes con HES/CEL. Para pacientes con HES/CEL con cinasa de fusión FIP1L1-PDGFR-alfa demostrada, se recomienda una dosis inicial de 100 mg diarios. Se puede considerar un aumento de dosis de 100 a 400 mg para estos pacientes en ausencia de reacciones adversas al fármaco si las valoraciones demuestran una respuesta insuficiente al tratamiento. Dosis en dermatofibrosarcoma protuberans (DFSP) irresecable, recurrente y/o metastásico: La dosis recomendada de imatinib es de 800 mg al día para pacientes con DFSP. Ajustes en la dosis por reacciones adversas: Reacciones adversas no hematológicas: Si una reacción adversa no hematológica severa se desarrolla con el uso de imatinib debe suspenderse el tratamiento hasta que se haya resuelto el evento. Subsecuentemente, puede reanudarse el tratamiento según sea adecuado, dependiendo de la severidad inicial del evento. Si ocurren elevaciones en bilirrubina mayores al triple del límite superior de las cifras institucionales normales (LINS) o si la concentración de transaminasas hepáticas asciende a más de 5 veces el LINS, se suspenderá la administración de imatinib hasta que los niveles de bilirrubina hayan regresado a ? 1.5 x el LINS y los niveles de transaminasas sean inferiores a 2.5 x el LINS. El tratamiento con imatinib puede luego proseguir con una dosis diaria reducida. En los adultos la dosis se debe disminuir de 400 a 300 mg o de 600 a 400 mg u 800 a 600 mg, y en los niños de 340 a 260 mg/m2/día. Reacciones adversas hematológicas: Se recomienda la reducción a la dosis o interrupción terapéutica para la neutrocitopenia y trombocitopenia severas conforme se indica en la siguiente tabla: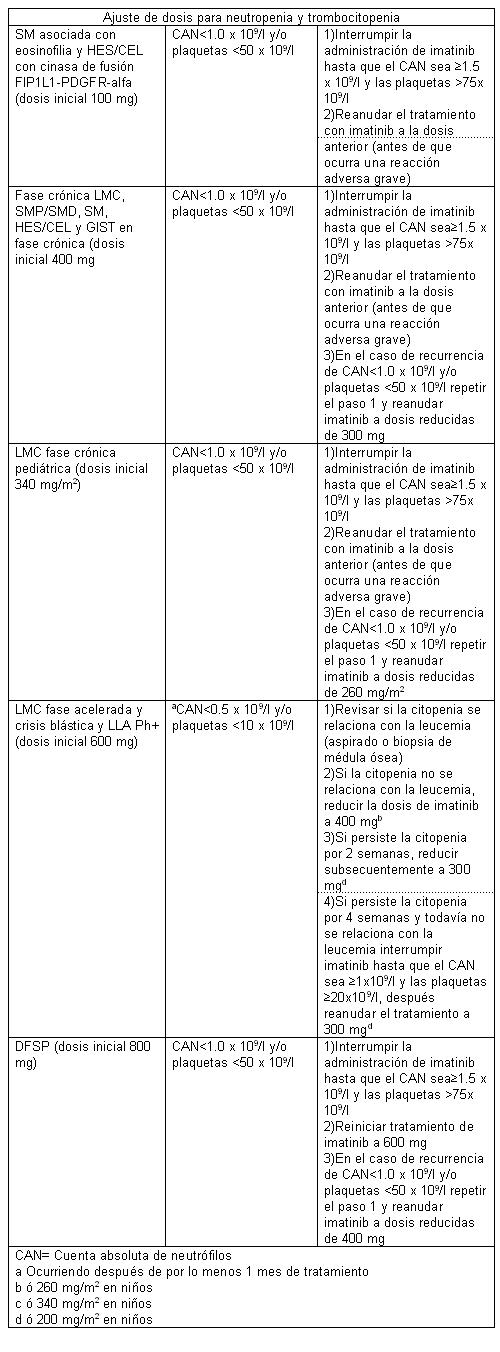
Niños: No hay experiencia con el uso de imatinib en niños con LMC menores de 2 años de edad. Existe experiencia muy limitada con el uso de imatinib en niños menores de 3 años de edad en otras indicaciones. Insuficiencia hepática: El imatinib se metaboliza principalmente en hígado. Los pacientes con una disfunción hepática leve, moderada o grave recibirán la dosis mínima recomendada de 400 mg al día, que se podrá reducir en caso de no ser tolerada (véase precauciones generales, reacciones secundarias y adversas, farmacocinética y farmacodinamia). Insuficiencia renal: El imatinib y sus metabolitos no son excretados en forma considerable por el riñón. Debido a que la depuración renal de imatinib es insignificante, no se espera una disminución en la depuración corporal total en pacientes con insuficiencia renal. Pacientes con insuficiencia renal media o moderada deben recibir la dosis mínima recomendada empezando con 400 mg por día. Aunque la información disponible es muy limitada (véase farmacocinética y farmacodinamia en humanos), los pacientes con insuficiencia renal severa o en diálisis, pueden también empezar con una dosis de 400 mg. Sin embargo se recomienda tener precaución con estos pacientes. La dosis puede reducirse si no es tolerada, o aumentarse por falta de efectividad (véase precauciones generales). Pacientes de edad avanzada: No se han observado diferencias farmacocinéticas importantes relacionadas con la edad en estudios clínicos que incluyeron a más del 20% de pacientes de 65 años de edad o más. No es necesaria una recomendación de dosis específica en pacientes de esa edad.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Se tienen datos escasos con la administración de dosis superiores a 800 mg. En caso de sobredosis, se debe observar al paciente y se deben de tomar medidas de apoyo que correspondan.
Presentación(es): Caja con 20 y 60 tabletas de 100 mg. Caja con 10 y 30 tabletas de 400 mg.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: No se deje al alcance de los niños. Su venta requiere receta médica. No se use en el embarazo ni lactancia. Este medicamento deberá ser administrado únicamente por médicos especialistas en hematología y oncología con experiencia en quimioterapia antineoplásica. Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa al correo farmacovigilancia@cofepris.gob.mx.
Nombre y domicilio del laboratorio: Hecho en México por: Laboratorios PiSA, S.A. de C.V. Av. Miguel Ángel de Quevedo No. 555. Col. Romero de Terreros, Delegación Coyoacán. C.P. 04310 D.F., México.
Número de registro del medicamento: 161M2014 SSA.