TARCEVA®
ROCHE
Denominación genérica: Erlotinib.
Forma farmacéutica y formulación: Comprimidos. Cada comprimido contiene: clorhidrato de erlotinib equivalente a 25 mg, 100 mg y 150 mg de erlotinib. Excipiente cbp un comprimido.
Indicaciones terapéuticas: Antineoplásico. Cáncer de pulmón de células no pequeñas (CPCNP): TARCEVA® está indicado para el tratamiento de los pacientes con cáncer de pulmón de células no pequeñas localmente avanzado o metastásico después del fracaso de al menos una línea de quimioterapia previa. Carcinoma pancreático: TARCEVA® en asociación con gemcitabina está indicado como tratamiento de primera línea de los pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico.
Farmacocinética y farmacodinamia: Farmacodinamia: propiedades y efectos: mecanismo de acción: el erlotinib inhibe intensamente la fosforilación intracelular de HER1/EGFR. HER1/EGFR se expresa en la superficie de células normales y células cancerosas. En modelos preclínicos, la inhibición de la EGFR-fosfotirosina produce estasis y muerte celular. Eficacia: cáncer de pulmón de células no pequeñas (CPCNP), (TARCEVA® en monoterapia): la eficacia y la seguridad de TARCEVA® se demostraron en un ensayo aleatorizado, a doble ciego y controlado con placebo (estudio BR,21). Este estudio se realizó en 17 países, en 731 pacientes con cáncer de pulmón de células no pequeñas localmente avanzado o metastásico después del fracaso de al menos una línea de quimioterapia previa. Los pacientes fueron aleatorizados en una proporción 2:1 para que recibieran TARCEVA® (150 mg) o placebo por vía oral una vez al día. Las variables de evaluación del estudio fueron la supervivencia general, el tiempo transcurrido hasta el deterioro de los síntomas relacionados con el cáncer pulmonar (tos, disnea y dolor), la tasa de respuesta, la duración de la respuesta, la supervivencia sin progresión y la seguridad. La variable principal de evaluación fue la supervivencia. Dado que la aleatorización se realizó en la proporción 2:1, 488 pacientes fueron asignados a TARCEVA® y 243 al placebo. No se seleccionó a los pacientes en función del estado HER1/EGFR, el sexo, la raza, los antecedentes de tabaquismo ni la clasificación histológica. Las características demográficas estaban bien equilibradas entre los dos grupos de tratamiento. Aproximadamente dos terceras partes de los pacientes eran varones. Aproximadamente un tercio presentaban un estado general inicial según la escala ECOG de 2 y el 9 % de 3. El 93% y el 92% de los pacientes de los grupos de TARCEVA® y placebo, respectivamente, habían recibido una línea previa que contenía platino, y el 36% y 37% de los pacientes, respectivamente, habían recibido tratamiento previo con taxanos. El 50% de los pacientes habían recibido sólo una línea de quimioterapia previa. Se evaluó la supervivencia en la población por intención de tratar. La mediana de la supervivencia general mejoró un 42,5% y fue de 6,7 meses en el grupo de TARCEVA® (IC del 95%: 5,5-7,8 meses) frente a 4,7 meses en el grupo del placebo (IC del 95%: 4,1-6,3 meses). El análisis principal de la supervivencia se ajustó en función de los factores de estratificación, como se registraron en el momento de la aleatorización (estado general según la escala ECOG, mejor respuesta al tratamiento previo, número de regímenes previos y exposición previa al platino), y en función del estado HER1/EGFR. En este análisis principal, la razón ajustada de riesgos instantáneos de muerte en el grupo de TARCEVA® en relación con el grupo del placebo fue de 0,73 (IC del 95%: 0,60-0,87) (p=0,001). El porcentaje de pacientes que vivían a los 12 meses fue del 31,2% y 21,5%, respectivamente. En la mayoría de los subgrupos se observó el efecto benéfico de TARCEVA® sobre la supervivencia. Para evaluar la robustez de los resultados de la supervivencia general, se examinaron en análisis unifactoriales exploradores una serie de subgrupos de pacientes formados por los valores de los factores de estratificación en el momento de la aleatorización y al inicio del estudio, el estado HER1/EGFR, la exposición previa a taxanos, los antecedentes de tabaquismo, el sexo, la edad, las características histológicas, el adelgazamiento previo, el tiempo transcurrido entre el diagnóstico inicial y la aleatorización y la localización geográfica. Casi todas las razones de riesgos instantáneos (HR, hazard ratio) en el grupo de TARCEVA® en comparación con el grupo del placebo fueron menores de 1,0 lo que sugiere que el benéfico de TARCEVA® sobre la supervivencia fue robusto en todos los subgrupos. Cabe señalar que el efecto benéfico de TARCEVA® sobre la supervivencia fue comparable en pacientes con un estado general inicial de 2-3 según la escala ECOG (HR=0,77) o un estado general de 0-1 (HR=0,73) y los pacientes que habían recibido una línea de quimioterapia (HR=0,76) o dos o más regímenes (HR=0,76). También se observó un efecto benéfico de TARCEVA® en los pacientes que no tuvieron una respuesta tumoral subjetiva (según los criterios RECIST). Esto se evidenció por la razón de riesgos instantáneos de muerte de 0,82 en los pacientes cuya mejor respuesta fue la enfermedad estable o la enfermedad progresiva. La tabla 1 resume los resultados del estudio, incluidos la supervivencia, el tiempo transcurrido hasta el deterioro de los síntomas relacionados con el cáncer de pulmón y la supervivencia sin progresión.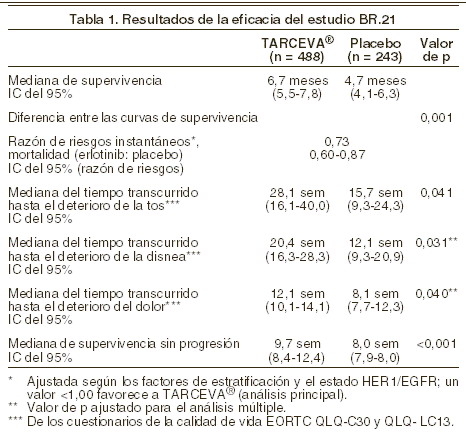
El deterioro de los síntomas se determinó utilizando los cuestionarios de la calidad de vida EORTC QLQ-C30 y QLQ-LC13. Las puntuaciones iniciales de la tos, la disnea y el dolor fueron similares en los dos grupos de tratamiento. TARCEVA® tuvo un efecto benéfico sobre los síntomas, prolongando significativamente el tiempo transcurrido hasta el deterioro de la tos (HR=0,75), la disnea (HR=0,72) y el dolor (HR=0,77), frente al placebo. Esta mejoría sobre los síntomas no se debió a un aumento del uso de radioterapia paliativa ni de medicación concomitante en el grupo de TARCEVA®. La mediana de la supervivencia sin progresión fue de 9,7 semanas en el grupo de TARCEVA® (IC del 95%: 8,4-12,4 semanas) en comparación con 8,0 semanas en el grupo del placebo (IC del 95%: 7,9-8,1 semanas). La razón de riesgos instantáneos de progresión, ajustada en función de factores de estratificación y del estado HER1/EGFR, fue de 0,61 (IC del 95%: 0,51-0,73) (p < 0,001). El porcentaje de supervivencia sin progresión a los 6 meses fue del 24,5% y el 9,3% respectivamente, para los grupos de TARCEVA® y de placebo. La tasa de respuesta objetiva según los criterios RECIST en el grupo de TARCEVA® fue del 8,9% (IC del 95%: 6,4-12,0%). La mediana de la duración de la respuesta fue de 34,3 semanas, oscilando entre 9,7 y 57,6 semanas. Se registraron dos respuestas (0,9%, IC del 95%: 0,1-3,4) en el grupo del placebo. La proporción de pacientes que mostraron una respuesta completa, una respuesta parcial o una enfermedad estable fue del 44,0% y el 27,5%, respectivamente, en los grupos de TARCEVA® y del placebo (p=0,004). Carcinoma pancreático (TARCEVA® administrado junto con gemcitabina): la eficacia y la seguridad de TARCEVA® en asociación con gemcitabina como tratamiento de primera línea se evaluó en un estudio aleatorizado, doble ciego y controlado con placebo (estudio PA.3), en 569 pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico. Se aleatorizaron 1:1 a los pacientes para recibir TARCEVA® (100 mg o 150 mg) o placebo una vez al día en una pauta continua junto con gemcitabina IV /1,000 mg/m2, ciclo 1: días 1, 8, 15, 22, 29, 36 y 43 de un ciclo de 8 semanas; ciclo 2 y ciclos siguientes; días 1, 8 y 15 de un ciclo de 4 semanas (dosis y pauta aprobadas para el carcinoma pancreático: revisar la información para prescribir de gemcitabina). TARCEVA® y placebo se tomaron por vía oral, una vez al día, hasta la progresión de la enfermedad o una toxicidad inaceptable. Las variables de evaluación del estudio fueron la supervivencia global, la tasa de respuesta y la supervivencia libre de progresión (SLP). También se examinó la duración de la respuesta. La variable principal de evaluación fue la supervivencia. Se aleatorizó a un total de 285 pacientes para recibir gemcitabina + TARCEVA® (261 pacientes en el grupo de 100 mg y 24 pacientes en el de 150 mg) y a 284 pacientes para recibir gemcitabina + placebo (260 pacientes en el grupo de 100 mg y 24 pacientes en el de 150 mg). En el grupo de 150 mg fueron muy pocas las observaciones para extraer conclusiones. Las características basales tanto demográficas como patológicas eran similares en los 2 grupos de tratamiento, 100 mg de TARCEVA® + gemcitabina o placebo + gemcitabina, salvo en la proporción ligeramente mayor de mujeres en el grupo de TARCEVA® (51%) que en el de placebo (44%). La mediana del tiempo entre el diagnóstico inicial y la aleatorización fue de aproximadamente 1,0 mes. Aproximadamente la mitad de los pacientes presentaban un estado general inicial según la escala ECOG de 1, y el 17%, de 2. La mayoría de los pacientes tenían metástasis al comienzo del estudio como manifestación inicial del cáncer de páncreas (77% en el grupo de TARCEVA® y 76% en el grupo del placebo). La supervivencia se evaluó en la población por intención de tratar a partir de los datos de supervivencia durante el seguimiento, que incluían 551 defunciones. Se presentan los resultados obtenidos en el grupo con la dosis de 100 mg (504 muertes). El cociente de riesgo instantáneo (hazard ratio) ajustado de fallecimiento en el grupo de TARCEVA® en comparación con el grupo del placebo fue de 0,82 (IC del 95%: 0,69 - 0,98) (p= 0,028). El porcentaje de los pacientes vivos a los 12 meses fue del 23,8% y el 19,4%, respectivamente, en los grupos de TARCEVA® y placebo. La mediana de supervivencia global fue de 6,4 y 4 meses, respectivamente, en los grupos de TARCEVA® y placebo.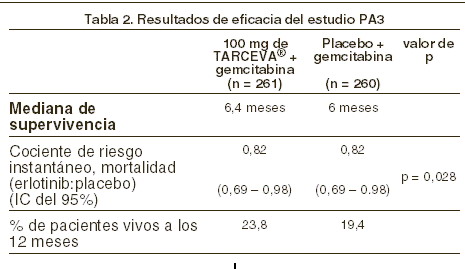
La mediana de SLP fue de 3,81 meses (16,5 semanas) en el grupo de TARCEVA® (IC del 95%: 3,58 - 4,93 meses), frente a 3,55 meses (15,2 semanas) en el grupo del placebo (IC del 95%: 3,29 - 3,75 meses) (p = 0,006). La mediana de duración de la respuesta fue de 23,9 semanas: entre 3,71 y 56 semanas. La tasa de respuesta objetiva (respuesta completa y respuesta parcial) fue del 8,6% y el 7,9%, respectivamente, en los grupos de TARCEVA® y placebo. La proporción de pacientes con respuesta completa, respuesta parcial o enfermedad estable fue del 59% y el 49,4%, respectivamente, en los grupos de TARCEVA® y placebo (p = 0,036). Farmacocinética. Absorción: el erlotinib se absorbe bien y tiene una amplia fase de absorción; las concentraciones plasmáticas máximas se alcanzaron 4 horas después de la administración oral. Un estudio en voluntarios sanos normales proporcionó una estimación de la biodisponibilidad del 59%. La comida puede aumentar la exposición después de una dosis oral. Tras la absorción, el erlotinib se une en gran medida en la sangre, fijándose cerca del 95% a los componentes sanguíneos, principalmente a las proteínas plasmáticas (esto es, la albúmina y la glucoproteína ácida 1), con una fracción libre del 5% aproximadamente. Distribución: el erlotinib tiene un volumen de distribución aparente medio de 232 l y se distribuye en los tejidos tumorales humanos. En un estudio de 4 pacientes (3 con cáncer de pulmón de células no pequeñas y 1 con cáncer de laringe) que recibieron TARCEVA® en dosis de 150 mg diarios por vía oral, las muestras tumorales obtenidas mediante escisión quirúrgica al día 9 de tratamiento revelaron concentraciones medias de erlotinib en el tumor de 1,185 ng/g de tejido. Esto corresponde a una media global del 63% de las concentraciones plasmáticas máximas observadas en estado de reposo. Los principales metabolitos activos estaban presentes en el tumor en concentraciones medias de 160 ng/g de tejido, lo que corresponde a una media global del 113% de las concentraciones plasmáticas máximas observadas en estado de reposo. Los estudios de distribución tisular utilizando un escaneo de cuerpo entero tras la administración oral de erlotinib marcado con [14C] en ratones atímicos con xenoinjertos de tumor HN5 han demostrado una distribución rápida y amplia, con concentraciones máximas del fármaco radiomarcado (aproximadamente el 73% de los valores en plasmas) observadas al cabo de 1 hora. Metabolismo: en el ser humano, el erlotinib es metabolizado por las enzimas hepáticas del sistema citocromo P450, principalmente por CYP3A4 y en menor grado por CYP1A2. Es posible que la metabolización extrahepática por CYP3A4 en el intestino, CYP1A1 en los pulmones y CYP1B1 en el tejido tumoral contribuya también a la depuración metabólica del erlotinib. Los estudios in vitro indican que aproximadamente el 80-95% del metabolismo del erlotinib corresponde a la enzima CYP3A4. Se han identificado tres vías metabólicas principales: 1) O-desmetilación de una cadena lateral o de ambas, seguida de la oxidación de los ácidos carboxílicos; 2) oxidación de la porción acetilénica seguida de la hidrólisis del ácido aril-carboxílico; y 3) hidroxilación aromática de la porción fenilacetilénica. Los metabolitos principales del erlotinib que se produjeron por O-desmetilación de alguna cadena lateral tienen una potencia comparable a la del erlotinib en los ensayos preclínicos in vitro y en los modelos tumorales in vivo. Están presentes en plasma en concentraciones que son < 10% de la concentración del erlotinib y muestran una farmacocinética similar a éste. Los metabolitos y cantidades mínimas del erlotinib se excretan predominantemente por las heces ( > 90%), eliminándose por el riñón sólo una pequeña cantidad de la dosis oral. Eliminación: depuración: un análisis farmacocinético de poblaciones en 591 pacientes que recibieron TARCEVA® en monoterapia reveló una depuración media aparente de 4,47 l/hora, con una mediana de la semivida de 36,2 horas. Por tanto, es previsible que el tiempo transcurrido hasta alcanzar concentraciones plasmáticas en estado de equilibrio sea de 7-8 días aproximadamente. No se observaron relaciones significativas entre la depuración aparente prevista y la edad, el peso, el sexo y la raza de los pacientes. Los factores del paciente que se correlacionan con la farmacocinética del erlotinib son la bilirrubina total en suero, las concentraciones de glucoproteína ácida 1 y el tabaquismo vigente. El aumento de las concentraciones séricas de la bilirrubina total y las concentraciones de glucoproteína ácida 1 se asociaron a una menor tasa de depuración del erlotinib. Los fumadores presentaron una tasa mayor de depuración del erlotinib (ver Interacciones medicamentosas y de otro género). Se realizó un segundo análisis de farmacocinética poblacional en el que se incluyeron los datos del erlotinib correspondientes a 204 pacientes con cáncer de páncreas tratados con erlotinib + gemcitabina. En este análisis se demostró que las covariables que afectaban la depuración del erlotinib en los pacientes del estudio del cáncer de páncreas eran muy similares a las observadas en el análisis de farmacocinética previo de la monoterapia. No se identificó ningún nuevo efecto de covariables. La coadministración de gemcitabina no tuvo ningún efecto en la depuración plasmática del erlotinib. Exposición: tras una dosis oral de 150 mg de TARCEVA®, en estado de equilibrio, la mediana del tiempo transcurrido hasta alcanzar la concentración plasmática máxima es aproximadamente de 4,0 horas, alcanzándose una mediana de las concentraciones plasmáticas máximas de 1.995 ng/ml. Antes de administrar la siguiente dosis a las 24 horas, la mediana de las concentraciones plasmáticas mínimas es de 1.238 ng/ml. La mediana del área bajo la curva (ABC) durante el intervalo entre dosis en estado de equilibrio es de 41.300 mcg.h/ml. Farmacocinética en poblaciones especiales: no se han realizado estudios específicos en niños ni en ancianos. Insuficiencia hepática: el erlotinib se elimina principalmente por el hígado. La exposición a erlotinib fue similar en pacientes con deterioro moderado de la función hepática (puntuación Child-Pugh 7-9) comparada con la exposición de pacientes con función hepática adecuada, incluyendo pacientes con cáncer de hígado primario o metástasis hepática. Insuficiencia renal: el erlotinib y sus metabolitos no se excretan en grado significativo por los riñones ( < 9% de una dosis única se excreta con la orina). No se han llevado a cabo estudios clínicos en pacientes con función renal disminuida. Fumadores: un estudio de farmacocinética en sujetos sanos no fumadores y fumadores de cigarrillos actualmente ha puesto de manifiesto que fumar cigarrillos eleva la depuración del erlotinib y reduce la exposición a éste. El ABC0-infinito en fumadores fue de 1/3 en comparación a la de ex/nunca fumadores (n=16 en cada uno de los brazos de fumadores y ex/nunca fumadores). Esta reducción en la exposición en los fumadores activos se debe presumiblemente a la inducción de CYP1A1 en pulmón y CYP1A2 en el hígado. En la Fase III del ensayo clínico clave de cáncer de pulmón de células no pequeñas (CPCNP), los fumadores actuales alcanzaron un estado constante de erlotinib a una concentración plasmática de 0,65 mg/ml (n=16) el cual fue, aproximadamente, dos veces menor que el de los ex-fumadores o el los pacientes que nunca han fumado (1,28 mg/ml, n=108). Este efecto se acompaña por un incremento del 24% en la depuración aparente en plasma de erlotinib. En la Fase I del estudio de escalamiento de dosis en pacientes con CPCNP en pacientes fumadores actuales los análisis farmacocinéticos a un estado constante indican un incremento proporcional a la dosis en la exposición a erlotinib cuando la dosis de TARCEVA® se incrementó de 150 mg a la dosis máxima tolerada de 300 mg. El estado constante de concentraciones en plasma a dosis de 300 mg en fumadores actuales en este estudio fue de 1,22 mg/ml (n=17) (ver Instrucciones especiales de dosificación e Interacciones medicamentosas y de otro género).
Contraindicaciones: TARCEVA® está contraindicado en pacientes con fuerte hipersensibilidad al erlotinib o a cualquier otro componente de TARCEVA®.
Precauciones generales: Se pueden producir interacciones farmacológicas clínicamente significativas con TARCEVA® (ver Interacciones medicamentosas y de otro género). Neumopatía intersticial: en raras ocasiones se han registrado casos de enfermedad pulmonar intersticial (EPI), algunos mortales, en pacientes que recibían TARCEVA® para el tratamiento del cáncer de pulmón de células no pequeñas, de cáncer de páncreas u otros tumores sólidos avanzados. En el estudio fundamental BR,21 en CPCNP, la incidencia de neumopatía intersticial grave fue del 0,8% tanto en el grupo del placebo como el grupo de TARCEVA®. En el estudio PA.3 sobre cáncer de páncreas en asociación con gemcitabina, la incidencia de episodios de alteraciones similares a la EPI fue del 2,5% en el grupo de TARCEVA® y gemcitabina, frente al 0,4% en el grupo con placebo y gemcitabina. La incidencia general en los pacientes tratados con TARCEVA® en todos los estudios (incluidos todos los estudios no controlados y los estudios con quimioterapia concomitante) es de aproximadamente el 0,6%. Algunos ejemplos de los diagnósticos registrados en pacientes en los que se sospechaba episodios de alteraciones similares a la EPI fueron: neumonitis, neumonitis por radiación, neumonitis por hipersensibilidad, neumonía intersticial, neumopatía intersticial, bronquiolitis obliterante, fibrosis pulmonar, síndrome de distrés respiratorio agudo, infiltración pulmonar y alveolitis. El comienzo de estos episodios de alteraciones similares a la EPI osciló entre algunos días y varios meses después de comenzado el tratamiento con TARCEVA®. La mayoría de los casos se asociaron a factores de confusión o factores contribuyentes, como quimioterapia concomitante o previa, radioterapia previa, neumopatía parenquimatosa preexistente, neumopatía metastática o infecciones pulmonares. En pacientes que desarrollen de forma aguda síntomas pulmonares inexplicados, nuevos o progresivos, como disnea, tos y fiebre, se debe interrumpir el tratamiento con TARCEVA® hasta la evaluación diagnóstica. Si se diagnostica una neumopatía intersticial, se suspenderá la administración de TARCEVA® e iniciará el tratamiento apropiado en función de las necesidades (ver Reacciones secundarias y adversas). Diarrea, deshidratación, desequilibrio electrolítico e insuficiencia renal: se ha producido diarrea en pacientes tratados con TARCEVA®, los casos moderados o graves de diarrea deben tratarse con loperamida. En ocasiones, puede ser necesario reducir la dosis. En caso de diarrea grave o persistente, náusea, anorexia o vómito asociados a deshidratación, se interrumpirá el tratamiento con TARCEVA® y se tomarán las medidas adecuadas para tratar la deshidratación (ver Reacciones secundarias y adversas). En raras ocasiones, se han notificado hipocalemia e insuficiencia renal (ocasionalmente mortal). Algunos reportes de insuficiencia renal fueron secundarios a deshidratación grave debido a diarrea, vómito y/o anorexia, mientras que otros fueron debidos a causas no relacionadas, sobre todo en pacientes que recibían quimioterapia al mismo tiempo. En los casos más graves o persistentes de diarrea, o conducentes a la deshidratación, particularmente en presencia de factores de riesgo agravantes (medicación concomitante, síntomas de enfermedad u otras condiciones predisponentes, la edad avanzada inclusive), se suspenderá la administración de TARCEVA® y se tomarán las medidas apropiadas para la rehidratación intensa de los pacientes por vía intravenosa. Además, deberán vigilarse la función renal y los electrólitos séricos, incluyendo el potasio en pacientes con riesgo de deshidratación. Hepatitis, insuficiencia hepática: durante el uso de TARCEVA® se han reportado casos raros de insuficiencia hepática (incluyendo fallecimientos). Los factores asociados o predisponentes incluyen enfermedad hepática preexistente o medicación hepatotóxica concomitante. Por lo tanto, en estos pacientes, se deben considerar pruebas de funcionamiento hepático periódicamente. Se debe interrumpir la administración de TARCEVA® si los cambios en la función hepática son graves (ver Reacciones secundarias y adversas). Insuficiencia hepática: la exposición a erlotinib fue similar en pacientes con deterioro moderado en la función hepática (puntuación Child-Pugh 7-9) comparada con la exposición de pacientes con función hepática adecuada, incluyendo pacientes con cáncer de hígado primario o metástasis hepática. No se han estudiado la seguridad ni la eficacia en pacientes con insuficiencia hepática. Efectos sobre la capacidad para conducir y utilizar máquinas: no se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas; ahora bien, el erlotinib no se ha asociado a alteraciones de la capacidad mental.
Restricciones de uso durante el embarazo y la lactancia: No existen estudios adecuados o bien controlados en mujeres embarazadas tratadas con TARCEVA®. Los estudios en animales han demostrado cierta toxicidad en la reproducción (ver Precauciones en relación con efectos de carcinogénesis, mutagenésis, teratogenésis y sobre la fertilidad). No se conocen los posibles riesgos para el ser humano. Se debe advertir a las mujeres con capacidad de procrear que eviten el embarazo mientras se hallen en tratamiento con TARCEVA®. Se deben utilizar métodos anticonceptivos adecuados durante el tratamiento y al menos las dos semanas siguientes a su conclusión. Sólo se mantendrá el tratamiento en mujeres embarazadas si el beneficio esperado para la madre supera los riesgos para el feto. Mujeres lactantes: no se sabe si el erlotinib se excreta con la leche materna. Dada la posibilidad de perjudicar al lactante, se debe advertir a las mujeres que no amamanten a sus hijos mientras reciben TARCEVA®. Insuficiencia hepática: la exposición a erlotinib fue similar en pacientes con deterioro moderado en la función hepática (puntuación Child-Pugh 7-9) comparada con la exposición de pacientes con función hepática adecuada, incluyendo pacientes con cáncer de hígado primario o metástasis hepática (ver Advertencias y Precauciones generales). No se han estudiado la seguridad ni la eficacia en pacientes con insuficiencia hepática.
Reacciones secundarias y adversas: Experiencia adquirida en los estudios clínicos: cáncer de pulmón de células no pequeñas (CPCNP). (TARCEVA® en monoterapia en el estudio BR,21): en un estudio aleatorizado y a doble ciego (BR,21) realizado en 17 países, 731 pacientes con cáncer de pulmón de células no pequeñas localmente avanzado o metastásico, tras el fracaso de al menos una línea de quimioterapia previa, fueron aleatorizados en la proporción 2:1 al tratamiento con TARCEVA® (150 mg) o a un grupo con placebo. El fármaco en estudio se administró diariamente por vía oral hasta la progresión de la enfermedad o hasta que se produjera una toxicidad inaceptable. Las erupciones cutáneas (rash) (75%) y la diarrea (54%) fueron los acontecimientos adversos más frecuentes, independientemente de la causalidad. En la mayoría de los pacientes, la intensidad era de grado 1 o 2 y se pudieron tratar sin intervención. El rash y la diarrea de grado 3 o 4 se produjeron en el 9% y el 6% respectivamente, de los pacientes tratados con TARCEVA®; cada uno de estos acontecimientos conllevó el abandono del estudio del 1% de los pacientes. Hubo que reducir la dosis por rash o diarrea en el 6% y el 1% de los pacientes, respectivamente. En el estudio BR,21, la mediana del tiempo transcurrido hasta el inicio del rash fue de 8 días y hasta el comienzo de la diarrea de 12 días. En la tabla 3 se enlistan, según el grado NCI-CTC (criterios comunes de toxicidad del Instituto Nacional del Cáncer de Canadá, por sus siglas en inglés), los eventos que fueron valorados por el patrocinador como reacciones adversas al medicamento atribuibles al tratamiento con TARCEVA®, y se señalan los acontecimientos adversos registrados con mayor frecuencia (≥3%) en los pacientes tratados con TARCEVA® que en el grupo del placebo en el estudio fundamental BR,21, y los que ocurrieron en ≥10% de los pacientes del grupo de TARCEVA®.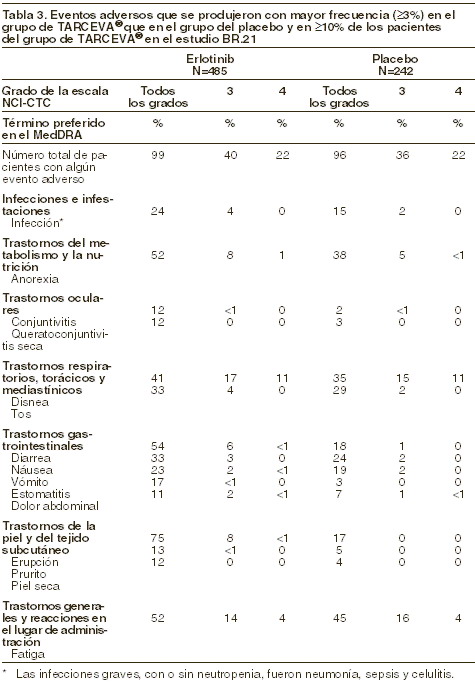
Carcinoma pancreático (TARCEVA® administrado junto con gemcitabina en el estudio PA.3): las reacciones adversas más frecuentes en el estudio fundamental PA.3 en pacientes con cáncer de páncreas tratados con TARCEVA® (100 mg) y gemcitabina fueron fatiga, erupción cutánea (rash) y diarrea. En el grupo de TARCEVA® y gemcitabina, se notificaron erupción y diarrea de grado 3 a 4 en el 5% de los pacientes. La mediana de la duración hasta el comienzo de la erupción y la diarrea fue de 10 y 15 días, respectivamente. A causa de la erupción y la diarrea, se redujo la dosis en el 2% de los pacientes y se suspendió la administración en hasta el 1% de los pacientes tratados con TARCEVA® y gemcitabina. En la cohorte (23 pacientes) del grupo con TARCEVA® (150 mg) y gemcitabina, hubo una tasa mayor de ciertas reacciones adversas específicas de clase, incluida la erupción, que requirieron con mayor frecuencia una reducción de la dosis o la retirada de la medicación. En la tabla 4 se encuentran resumidas las reacciones adversas reportadas con una frecuencia mayor (≥3%) en los pacientes tratados con TARCEVA® (100 mg) y gemcitabina que en el grupo del placebo y gemcitabina en el estudio fundamental PA.3 y en al menos el 10% de los pacientes del grupo de TARCEVA® (100 mg) y gemcitabina, según la escala NCI-CTC (criterios comunes de toxicidad del Instituto Nacional del Cáncer de Canadá, por sus siglas en inglés). Los eventos enlistados fueron valorados por el patrocinador como reacciones adversas al medicamento atribuibles al tratamiento con TARCEVA®.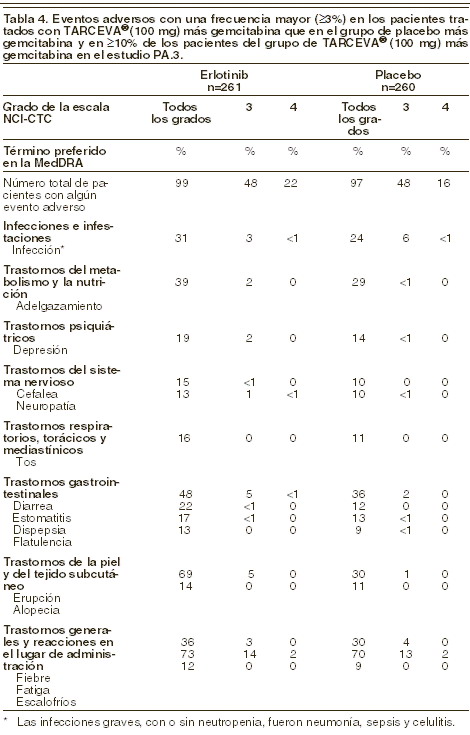
Otras observaciones (basadas en los datos de todos los estudios clínicos): la población principal de seguridad de TARCEVA® fue definida en los 800 pacientes que recibieron al menos una dosis de 150 mg de TARCEVA® en monoterapia y más de 300 pacientes que recibieron TARCEVA® 100 o 150 mg en combinación con gemcitabina. Las reacciones adversas señaladas a continuación se observaron en los pacientes tratados con 150 mg de TARCEVA® como monoterapia o con 100 mg en combinación con gemcitabina. Se utilizaron los siguientes términos para clasificar los efectos indeseables según la frecuencia: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100, < 1/10), infrecuentes (≥ 1/1.000, < 1/100), raros (≥ 1/10.000, < 1/1.000); muy raros ( < 1/10.000), incluidos los informes aislados. En las tablas 3 y 4 se presentan las reacciones adversas más comunes. A continuación se resumen otras reacciones adversas. Trastornos gastrointestinales: en los estudios clínicos se han notificado con frecuencia casos de hemorragia gastrointestinal, algunos asociados con la administración simultánea de warfarina (ver Interacciones medicamentosas y de otro género) y otros con la de administración de AINEs. Trastornos hepatobiliares: en los ensayos clínicos de TARCEVA®, han sido frecuentes las anomalías en las pruebas de función hepática (incluido el aumento de ALT, AST y bilirrubina). En el estudio PA.3, fueron muy frecuentes. En la mayoría de los casos, fueron de intensidad leve o moderada, de naturaleza transitoria o asociadas a metástasis hepáticas. Durante el uso de TARCEVA®, se han reportado casos raros de insuficiencia hepática (incluyendo fallecimientos). Los factores asociados o predisponentes incluyen enfermedad hepática preexistente o medicación hepatotóxica concomitante (ver Precauciones generales). Trastornos oculares: se han registrado frecuentemente queratitis en los ensayos clínicos de TARCEVA®. La conjuntivitis ha sido frecuente en los estudios de cáncer de páncreas. En pacientes que reciben TARCEVA®, raramente se han reportado ulceraciones corneales, como una complicación de la inflamación mucocutánea. Trastornos respiratorios, torácicos y mediastínicos: han sido infrecuentes los casos de episodios similares a la EPI (incluidos los mortales) en pacientes que recibían TARCEVA® como tratamiento del cáncer pulmonar de células no pequeñas y otros tumores sólidos avanzados (ver Precauciones generales). Se han notificado frecuentemente casos de epistaxis en los estudios tanto de CPCNP como de carcinoma pancreático. Trastornos de la piel y del tejido subcutáneo: la piel reseca se ha observado frecuentemente en estudios clínicos de cáncer de páncreas. La alopecia ha sido una reacción frecuente en los estudios de CPCNP. En estudios clínicos se han reportado cambios en cabellos y uñas, en su mayoría no serios, p. ej. se reportó paroniquia como común; se han reportado hirsutismo, cambios en pestañas y cejas, y uñas frágiles o sueltas, como poco comunes.
Interacciones medicamentosas y de otro género: En el ser humano, el erlotinib es metabolizado en el hígado por los citocromos hepáticos, principalmente CYP3A4 y, en menor grado CYP1A2, y por la isoforma pulmonar CYP1A1. Pueden producirse interacciones con fármacos inhibidores o inductores de estas enzimas o que sean metabolizados por ellas. Los inhibidores potentes de la actividad de CYP3A4 reducen el metabolismo del erlotinib y aumentan su concentración plasmática. La inhibición del metabolismo de CYP3A4 por el ketoconazol (200 mg p.o. 2 veces/día durante 5 días) produjo un aumento de la exposición al erlotinib (86% en la mediana de la exposición al erlotinib [ABC]) y un aumento del 69% en comparación con el erlotinib solo. Cuando se coadministró TARCEVA® con ciprofloxacino, un inhibidor de ambos CYP3A4 y CYP1A2, la exposición a erlotinib (ABC) y concentración máxima (Cmáx) incrementaron 39% y 17%, respectivamente Por tanto, la administración de TARCEVA® junto con inhibidores potentes de CYP3A4 o CYP3A4/CYP1A2 combinados exige precaución. En estas situaciones, se debe reducir la dosis de TARCEVA® si se observan signos de toxicidad. Los inductores potentes de la actividad de CYP3A4 aumentan el metabolismo del erlotinib y disminuyen significativamente su concentración plasmática. La inducción del metabolismo de CYP3A4 por la rifampicina (600 mg/día p.o. durante 7 días) produjo una reducción del 69% en la mediana del ABC del erlotinib posterior a una dosis de 150 mg de TARCEVA®, en comparación con la monoterapia con TARCEVA®. El pretratamiento y coadministración de rifampicina con una dosis única de 450 mg de TARCEVA® dio lugar a una exposición media de erlotinib (ABC) de 57,5% de la dosis única de 150 mg de TARCEVA® en ausencia de tratamiento con rifampicina. Cuando sea posible, se deben considerar tratamientos alternos que no tengan una potente actividad inductora de CYP3A4. Para aquellos pacientes que requieran tratamiento con TARCEVA® de forma concomitante a un inductor potente de CYP3A4, tal como la rifampicina, se debe considerar un incremento en la dosis a 300 mg siempre y cuando se monitoree cuidadosamente su seguridad, y si es bien tolerado por más de 2 semanas; se debe considerar un incremento posterior hasta 450 mg con vigilancia estrecha de seguridad. Dosis mayores no se han estudiado bajo este escenario. El pretratamiento o la coadministración de TARCEVA® no alteró la depuración de los sustratos prototípicos de CYP3A4, midazolam y eritromicina. Por ello, es improbable que haya una interacción significativa con otros sustratos de CYP3A4. La disponibilidad oral de midazolam parece decrecer hasta en 24%, lo cual no estuvo atribuido a los efectos de la actividad de CYP3A4. La solubilidad de erlotinib es dependiente del pH. La solubilidad de erlotinib disminuye cuando el pH aumenta. Los medicamentos que modifican el pH del tracto gastrointestinal superior pueden alterar la solubilidad del erlotinib y por lo tanto su biodisponibilidad. La co-administración de TARCEVA® con omeprazol, un inhibidor de la bomba de protones, disminuyó la exposición de erlotinib (ABC) y la (Cmáx) en 46% y 61% respectivamente. No hubo cambios en la vida media o Tmax. La administración concomitante de TARCEVA® con ranitidina a dosis de 300 mg, un antagonista del receptor H2 dio lugar a un decremento de la exposición de erlotinib (ABC) y su concentración máxima en un 33% y un 54% respectivamente. La administración concomitante con medicamentos que reducen la producción de secreción ácida gástrica deben ser evitados en el paciente manejado con TARCEVA®. Es posible que el incrementar la dosis de TARCEVA® cuando se coadministre con estos agentes no compense la concentración por esta pérdida de exposición. Sin embargo si la administración de TARCEVA® se realiza 2 h antes o 10 h después de la administración de ranitidina a 150 mg cada 12 h, la exposición a erlotinib decrece únicamente en un 15% y la Cmáx en un 17%. Si los pacientes requieren ser manejados con un antagonista H2, entonces se debe considerar este esquema de manejo. TARCEVA® debe ser tomado 2 h antes o 10 h después de la ingesta de antagonistas H2. En estudios clínicos se han registrado elevaciones del índice internacional normalizado (INR) y acontecimientos hemorrágicos, como hemorragia digestiva, (ver Reacciones secundarias y adversas), asociados a la administración concomitante de warfarina. Se deben realizar controles periódicos de los pacientes que reciben warfarina u otros anticoagulantes derivados cumarínicos para detectar cambios en el tiempo de protrombina o el INR. Se debe advertir a los fumadores que deben dejar de fumar, ya que se sabe que fumar un cigarro induce CYP1A1 y CYP1A2, lo que disminuye la exposición a erlotinib en 50-60% (ver Dosis y vía de administración y Farmacocinética en poblaciones especiales). En un estudio de fase Ib, no se detectó ningún efecto significativo de la gemcitabina en la farmacocinética del erlotinib, ni viceversa, del erlotinib en la farmacocinética de la gemcitabina.
Alteraciones en los resultados de pruebas de laboratorio: Los efectos de la administración a largo plazo observados al menos en una especie animal o estudio incluyeron disminución de los parámetros eritrocitarios y aumento de los leucocitos, principalmente los neutrófilos. Se produjeron aumentos relacionados con el tratamiento de las concentraciones de alanina-aminotransferasa (ALT), aspartato-aminotransferasa (AST) y bilirrubina. Los estudios en perros no evidenciaron una prolongación del segmento QT. En una revisión sistemática centralizada de los datos electrocardiográficos de 152 individuos de siete estudios con voluntarios sanos, no se hallaron signos de prolongación del segmento QT, como tampoco se han encontrado en los estudios clínicos signos de arritmias asociados a la prolongación del segmento QT. Postcomercialización: trastornos de la piel y tejido subcutáneo: cambios en el cabello y las uñas, principalmente no serios, se reportaron como poco comunes durante la vigilancia postcomercialización, por ej. hirsutismo, cambios en pestañas/cejas, paroniquia y uñas débiles y quebradizas.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: en estudios preclínicos no se observó un potencial carcinogénico. El erlotinib no fue genotóxico ni clastogénico en estudios de toxicidad genética. Se han iniciado estudios de carcinogenicidad a largo plazo en ratas y ratones, sin embargo, no se han observado lesiones premalignas proliferativas en estudios de toxicidad crónica hasta los 6 meses. Mutagenicidad: el erlotinib es negativo en la batería estándar de ensayos de genotoxicidad. Bloqueo de la fertilidad: no se observó bloqueo de la fertilidad en estudios con ratas macho y hembra en dosis cercanas a los niveles de dosis máxima tolerada (DMT). Teratogenicidad: los datos toxicológicos reproductivos en ratas y conejos indican que posterior a la exposición a erlotinib a dosis cercanas a la DMT y/o dosis que fueron maternalmente tóxicas, ocurrió embriotoxicidad, pero no hubo evidencia de teratogenicidad o desarrollo teratogénico o anormal pre o postnatal. La toxicidad materna tanto en ratas y conejos en estos estudios ocurrió a niveles plasmáticos de exposición simi