TIVICAY
GSK
Denominación genérica: Dolutegravir (DTG).
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene 50 mg de dolutegravir (como dolutegravir sódico).
Indicaciones terapéuticas: Tratamiento contra la infección del virus de la inmunodeficiencia humana (VIH-1) en combinación con otros agentes antirretrovirales en adultos y niños de más de 12 años de edad.
Farmacocinética y farmacodinamia: Farmacocinética: La farmacocinética de TIVICAY® es similar entre sujetos sanos y sujetos infectados con VIH. La variabilidad PK de dolutegravir es de baja a moderada. En los estudios de Fase 1 en sujetos sanos, la CVb% entre sujetos para el AUC y la Cmax, varió de ~20 a 40% y la Ct de 30 a 65% entre estudios. La variabilidad PK entre sujetos de DTG fue más alta en sujetos infectados con VIH en comparación con sujetos sanos. La variabilidad dentro de los sujetos (CVw%) es menor que la variabilidad entre sujetos. Absorción: TIVICAY® experimenta una rápida absorción tras la administración oral, con una mediana de Tmax a las 2 a 3 horas posteriores a la administración de la dosis para la formación en tabletas. La linealidad de la farmacocinética de dolutegravir es dependiente de la dosis y la formulación. Tras la administración oral de las formulaciones en tabletas, en general TIVICAY® mostró una farmacocinética no lineal con aumentos menos que proporcionales a la dosis en la exposición plasmática de 2 a 100 mg; sin embargo el aumento en la exposición a dolutegravir parece ser proporcional a la dosis de 25 mg a 50 mg. TIVICAY® puede administrarse con o sin alimentos. Los alimentos aumentaron el grado y disminuyeron la velocidad de absorción de dolutegravir. La biodisponibilidad de dolutegravir depende del contenido de alimento: las comidas bajas, moderadas y altas en grasas aumentaron el AUC (0-t) de dolutegravir en 33%, 41% y 66%, aumentaron la Cmax en 46%, 52% y 67%, prolongaron la Tmax a 3, 4 y 5 horas de 2 horas en condiciones de ayuno, respectivamente. Estos aumentos no son clínicamente significativos. Aún no se establece la biodisponibilidad absoluta de dolutegravir. Distribución: Dolutegravir experimenta un alto grado de unión (aproximadamente 99.3%) a proteínas plasmáticas humanas, con base en datos in vitro. Se estima que el volumen aparente de distribución (después de la administración oral de la formulación en suspensión, Vd/F) es de 12.5 L. La unión de dolutegravir a proteínas plasmáticas fue independiente de la concentración. Los cocientes de concentración de radioactividad relacionada con el fármaco en plasma y sangre entera se promedian en 0.441 a 0.535, lo cual indica una asociación mínima de radioactividad con los componentes celulares de la sangre. Se estima que la fracción libre de dolutegravir en plasma es 0.2 a 1.1 % en sujetos sanos, ~0.4 a 0.5% en sujetos con insuficiencia hepática moderada y 0.8 a 1.0% en sujetos con insuficiencia renal grave, y de 0.5% en pacientes infectados con VIH-1. Dolutegravir se concentra en el líquido cefalorraquídeo (LCR). En 12 sujetos vírgenes a tratamiento recibiendo un régimen de dolutegravir mas abacavir/lamivudina (3TC) por 16 semanas, las concentraciones en LCR de dolutegravir promediaron 15.4 ng/mL a la semana 2 y 12.6 ng/mL a la semana 16, fluctuando de 3.7 a 23.2 ng/mL (comparable a la concentración plasmática no unida). La relación LCR: plasma de las concentración de DTG fluctuó de 0.11 a 2.04%. Las concentraciones de dolutegravir en LCR excedieron la IC50, apoyando la mediana de disminución desde la basal en el LCR del RNA de VIH-1 de 2.2 log después de 2 semanas de tratamiento y 3.4 log después de 16 semanas (ver Farmacodinámica). Dolutegravir está presente en el tracto genital de machos y hembras. El AUC en líquido cervicovaginal, tejido cervical, y tejido vaginal, fue de 6 a 10% al correspondiente en plasma en estado de equilibrio. El AUC fue de 7% en semen y de 17% en tejido rectal, en comparación con los valores correspondientes en plasma en estado de equilibrio. Metabolismo: Dolutegravir se metaboliza principalmente a través de UGT1A1, con un menor componente de CYP3A (9.7% de la dosis total administrada en un estudio de equilibrio de masa humana). Dolutegravir es el compuesto predominante que circula en el plasma, y la eliminación renal de fármaco inalterado es baja ( < 1% de la dosis). Cincuenta y tres por ciento de la dosis oral total se excreta de manera inalterada en las heces. Se desconoce si la totalidad o parte de esto se debe al fármaco no absorbido o a la excreción biliar del conjugado glucurónido, que puede ser degradado aún más para formar el compuesto principal en la luz del intestino. Treinta y uno por ciento de la dosis oral total se excreta en la orina, representada por el glucurónido de éter de dolutegravir (18.9% de la dosis total), el metabolito N- desalquilación (3.6% de la dosis total), y un metabolito formado por oxidación en el carbono bencílico (3.0% de la dosis total). Eliminación: Dolutegravir tiene una vida media terminal de ~14 horas y una depuración aparente (CL/F) de 0.56 L/hr. Población de pacientes especiales: Niños: En un estudio pediátrico incluyendo 23 niños infectados con VIH-1 previamente tratados con antirretrovirales y adolescentes de 12 a 18 años de edad, se evaluó la farmacocinética de dolutegravir en 10 niños y mostró que la dosis de TIVICAY® de 50 mg una vez al día, resultó en una exposición a dolutegravir en sujetos pediátricos comparable a la observada en adultos que recibieron TIVICAY® 50 mg una vez al día (Tabla 1).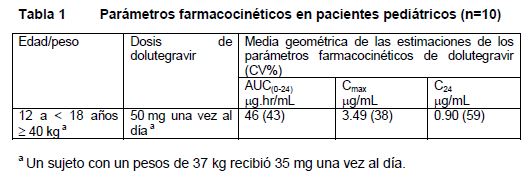
Ancianos: El análisis farmacocinético poblacional de dolutegravir utilizando datos de adultos infectados con VIH-1 mostró que no se observó un efecto relevante de la edad sobre la exposición a dolutegravir. Los datos farmacocinéticos de dolutegravir en sujetos > 65 años de edad son limitados. Insuficiencia renal: La eliminación renal del fármaco sin cambios es una vía menor de eliminación de dolutegravir. Se realizó un estudio de la farmacocinética de dolutegravir en sujetos con insuficiencia renal severa (CrCl < 30 mL/min). No se observaron diferencias clínicamente importantes entre sujetos con insuficiencia renal severa (CrCl < 30mL/min) y sujetos sanos con características similares. No es necesario ajustar la dosis en pacientes con insuficiencia renal. Dolutegravir no ha sido estudiado en pacientes sometidos a diálisis, aunque no se esperan diferencias en la exposición. Insuficiencia hepática: Dolutegravir se metaboliza y elimina principalmente en el hígado. En un estudio que comparó 8 sujetos con insuficiencia hepática moderada (categoría Child Pugh B) con 8 controles adultos sanos con características similares, la exposición a una dosis única de 50 mg de dolutegravir, fue similar entre ambos grupos. No es necesario ajustar la dosis en pacientes con insuficiencia hepática de leve a moderada. No se ha estudiado el efecto de la insuficiencia hepática severa sobre la farmacocinética de dolutegravir. Polimorfismo en las enzimas metabolizadas de fármacos: No existe evidencia de que los polimorfismos comunes en las enzimas metabolizantes de fármacos, alteren la farmacocinética de dolutegravir en un grado clínicamente significativo. En un meta-análisis que hizo uso de muestras farmacogenómicas recolectadas en estudios clínicos realizados en sujetos sanos, los sujetos con genotipos UGT1A1 (n= 7) que confieren un mal metabolismo de dolutegravir, mostraron una eliminación 32% menor de dolutegravir, y 46% mostraron AUC más altas en comparación con sujetos con genotipos asociados con un metabolismo UGT1A1 normal (n= 41). Los polimorfismos CYP3A4, CYP3A5, y NR1I2 no se asociaron con diferencias en la farmacocinética de dolutegravir. Género: El grado de exposición a dolutegravir en sujetos sanos parece ser ligeramente superior (~20%) en mujeres que en hombres, con base en los datos obtenidos en un estudio de sujetos sanos (hombres n=17, mujeres n=24). Los análisis PK poblacionales que hicieron uso de datos farmacocinéticos agrupados de estudios en adultos en Fase IIb y III no revelaron algún efecto clínicamente relevante del género en el grado de exposición a dolutegravir. Raza: Los análisis PK poblacionales que hicieron uso de datos farmacocinéticos agrupados de estudios en adultos en Fase IIb y III no revelaron algún efecto clínicamente relevante de la raza en el grado de exposición a dolutegravir. La Farmacocinética de dolutegravir después de la administración oral de una dosis única en sujetos Japoneses, parece ser similar a los parámetros observados en sujetos Occidentales (EE.UU.). Coinfección con Hepatitis B o C: El análisis farmacocinético poblacional indicó que la coinfección por virus de la hepatitis C no tuvo un efecto clínicamente relevante en el grado de exposición a dolutegravir. Existen datos limitados en sujetos con coinfección por hepatitis B. Farmacodinamia: Mecanismo de acción: TIVICAY® inhibe la unión de la integrasa del VIH mediante la unión al sitio activo de la integrasa y a través del bloqueo del paso de transferencia de cadena e integración del ácido desoxirribonucleico (ADN) retroviral, la cual es esencial para el ciclo de replicación del VIH. Los estudios bioquímicos de transferencia de cadenas usando substrato purificado de integrasa VIH 1 y DNA pre-procesado resultó en valores IC50 de 2.7 nM y 12.6 nM. In vitro, dolutegravir se disocia lentamente del sitio activo del complejo integrasa-DNA salvaje (t ½ 71 horas). Efectos farmacodinámicos: En un estudio aleatorizado de rango de dosis, los sujetos infectados con VIH 1 tratados con monoterapia con TIVICAY®, mostraron una actividad antiviral rápida y dependiente de la dosis, con disminuciones medias desde la basal el día 11 del RNA de VIH-1 de 1.5, 2.0, y 2.5 log10 para dolutegravir 2 mg, 10 mg, y 50 mg una vez al día, respectivamente. Esta respuesta antiviral se mantuvo durante 3 a 4 días después de la última dosis en el grupo de 50 mg. Actividad antiviral en cultivo celular: Las células mononucleares de sangre periférica (PBMC) infectadas con la cepa BaL de VIH-1 o la cepa NL432 de VIH-1 dio lugar a IC50 de dolutegravir de 0.51 nM y 0.53 nM, respectivamente. Las células MT-4 infectadas con la cepa IIIB de VIH -1 e incubadas con dolutegravir durante 4 o 5 días dieron lugar a IC50 de 0.71 y 2.1 nM. En un ensayo de susceptibilidad de la integrasa viral que hizo uso de la región de codificación de la integrasa de 13 aislados de clado B clínicamente diversos, dolutegravir demostró una potencia antiviral similar a las cepas de laboratorio, con una media de IC50 de 0.52 nM. Cuando se realizaron análisis en ensayos de células mononucleares de sangre periférica (PBMC) contra un panel que consistía en 24 aislados clínicos de VIH-1 [grupo M (clado A, B, C, D, E, F y G) y grupo O] y 3 aislados clínicos de VIH -2, la media geométrica de IC50 fue de 0.20 nM y los valores de IC50 oscilaron entre 0.02 y 2.14 nM para los aislados de VIH-1, mientras que la media geométrica de IC50 fue de 0.18 nM y los valores de IC50 oscilaron entre 0.09 y 0.61 nM para los aislados de VIH-2. Actividad antiviral en combinación con otros agentes antivirales: Ningún fármaco con actividad anti-VIH inherente fue antagonista con dolutegravir, o si no tenía una actividad anti-VIH inherente (ribavirina), no tuvo algún efecto aparente en la actividad de dolutegravir. La actividad anti-VIH-1 in vitro de dolutegravir fue estudiada en un formato cuadriculado en combinación con fármacos anti-VIH representativos (estavudina, abacavir, efavirenz, nevirapina, lopinavir, amprenavir, enfuvirtida, maraviroc y raltegravir), Efecto de las proteínas sericas y las proteínas séricas humanas: Los estudios in vitro sugirieron un cambio de 75 veces en la IC50 de dolutegravir en presencia de suero humano al 100% (por el método de extrapolación), y se estimó que la IC90 ajustada a proteínas (PA-IC90) en PBMC era de 64 ng/mL. La concentración valle de dolutegravir para una una dosis única de 50 mg en sujetos sin tratamiento previo con inhibidores de la integrasa fue de 1.20m g/mL, 19 veces superior a la PA-IC90 estimada. Resistencia in vitro: Aislado de VIH-1 de tipo salvaje: No se observaron virus altamente resistentes a dolutegravir durante el paso de 112 días de la cepa IIIB, con un cambio máximo de 4.1 veces (FC) observado para las poblaciones de virus resistentes que pasaron con sustituciones en las posiciones de IN conservadas, S153Y y S153F. Paso de la cepa NL432 de VIH-1 de tipo salvaje en presencia de dolutegravir seleccionado para sustituciones de E92Q (FC=3.1 para virus de población de paso) y G193E (FC=3.2 para virus de población de paso) en el Día 56. Paso adicional de virus de tipo salvaje subtipos B, C, y A/G en presencia de dolutegravir seleccionado para R263K, G118R, y S153T. Actividad anti-VIH contra cepas resistentes: Cepas resistentes a inhibidores de la transcriptasa inversa e inhibidores de la proteasa: Dolutegravir demostró una potencia equivalente contra 2 clones mutantes de VIH-1 resistentes a RTI no nucleosídico (NN), 3 resistentes a RTI nucleosídico (N) y 2 resistentes a PI (1 triple y 1 séxtuple), en comparación con la cepa de tipo salvaje. Cepas de VIH-1 resistentes a inhibidores de la integrasa: Se produjeron 60 virus VIH-1 mutantes resistentes a inhibidores de la integrasa (28 con sustituciones únicas y 32 con 2 o más sustituciones) a partir del virus de tipo salvaje NL-432 utilizando mutagénesis dirigida a sitio. Dolutegravir mostró una actividad anti-VIH (susceptibilidad) con un FC < 5 contra 27 de 28 de los virus mutantes resistentes a inhibidores de la integrasa con sustituciones únicas, incluyendo T66A/I/K, E92Q/V, Y143C/H/R, Q148H/K/R y N155H, mientras que para raltegravir y elvitegravir hubo 17/28 y 11/21 virus mutantes con un FC < 5, respectivamente. Además, de los 32 virus mutantes resistentes a inhibidores de la integrasa con 2 o más sustituciones, 23 de 32 mostraron un FC < 5 a dolutegravir, en comparación con un FC < 5 para 4 de 32 para raltegravir y FC < 5 para 2 de 25 para elvitegravir. Cepas de VIH-2 resistentes a inhibidores de la integrasa: Se construyeron virus VIH-2 mutantes dirigidos a sitio basándose en sujetos infectados con VIH-2 y tratados con raltegravir que mostraron una falla virológica. En general, los FC de VIH-2 observados fueron similares a los de VIH-1 con mutaciones de la vía similares El FC fue de < 5 contra 4 virus VIH-2; S163D, G140A/Q148R, A153G/N155H/S163G y E92Q/T97A/N155H/S163D; para E92Q/N155H, FC = 8.5 para dolutegravir, y para G140S/Q148R FC = 17 para dolutegravir. Dolutegravir, raltegravir y elvitegravir tuvieron la misma actividad contra el virus VIH-2 mutante dirigido a sitio con S163D como tipo salvaje, y por el resto de los virus VIH-2 mutantes, los rangos de FC para raltegravir fueron de 6.4 a 420 y los rangos de FC para elvitegravir eran de 22 a 640. Aislados clínicos de sujetos con falla virológica bajo tratamiento con raltegravir: Se examinaron treinta muestras de aislados clínicos con resistencia genotípica y fenotípica a raltegravir (FC mediana > 81) en cuanto a susceptibilidad a dolutegravir (FC mediana de 1.5) utilizando el ensayo de Monogram Biosciences PhenoSense. El FC medio a dolutegravir para aislados que contienen cambios en G140S + Q148H fue de 3.75; en G140S + Q148R fue de 13.3; en T97A + Y143R fue de 1.05 y en N155H fue de 1.37. Setecientos cinco cepas resistentes a raltegravir de pacientes tratados previamente con raltegravir fueron analizadas en cuanto a susceptibilidad a dolutegravir utilizando el ensayo de Monogram Biosciences PhenoSense. Dolutegravir tiene un FC < menor que o igual a 10 contra 93.9% de los 705 aislados clínicos, cabe destacar que 16 (9%) de los 184 aislados con Q148 + 1 sustitución de resistencia INSTI y 25 (27%) de los 92 aislados clínicos con Q148 + ≥ 2 sustituciones de resistencia INSTI tuvieron un cambio de más de 10 veces. Resistencia in vivo: pacientes sin tratamiento previo con inhibidores de la integrasa: No se aislaron mutaciones resistentes a INI ni resistencias emergentes con el tratamiento con un NRTI de base con TIVICAY® 50 mg una vez al día en estudios en pacientes vírgenes a tratamiento (estudios SPRING-1, SPRING-2, SINGLE y FLAMINGO). En el estudio SAILING realizado en pacientes con tratamiento previo (y vírgenes a integrasas) (n= 354 en el grupo de dolutegravir), a la semana 48 se observaron sustituciones a integrasas emergentes con el tratamiento en 4 de los 17 sujetos recibiendo dolutegravir con falla virológica. De esos cuatro, 2 sujetos tuvieron una sustitución única de integrasa en R263K, con un FC máximo de 1.93, un sujeto tuvo una sustitución polimórfica de integrasa V151V/I, con Fc máxima de 0.92, y 1 sujeto tuvo mutaciones pre-existentes de integrasa y se asume que había tenido contacto con integrasa o se infectó por transmisión con virus resistente a los inhibidores de la integrasa (ver Estudios clínicos). Resistencia in vivo: pacientes resistentes a inhibidores de la integrasa: El estudio VIKING-3 examinó TIVICAY® (más un tratamiento optimizado de base) en sujetos con resistencia pre existente a INI. Treinta y seis sujetos (36/183) presentaron falla virológica definida por el protocolo hasta la Semana 24. De estos, 32 tenían datos pareados de resistencia basal y PDVF para el análisis, y 17/32 (53%) tenían mutaciones emergentes con el tratamiento. Las mutaciones emergentes con el tratamiento o las mezclas de mutaciones observadas fueron L74L/m (n=1), E92Q (n= 2), T97A (n= 9), E138K/A (n= 8), G140S (n= 2), Y143H (n= 1), S147G (n= 1), Q148H/K/R (n= 4), y N155H (n= 1) y E157E/Q (n=1). Catorce de los 17 sujetos con virus que presentaban mutaciones emergentes con el tratamiento, portaban virus de la vía Q148 en la basal o previamente. Cinco sujetos más experimentaron PDVF entre las semanas 24 y 48, y 2 de estos 5 tuvieron mutaciones emergentes con el tratamiento. Las mutaciones emergentes con el tratamiento o mezclas de mutaciones observadas fueron L741 (n=1), N155H (n=2). Efectos en el electrocardiograma: En un estudio aleatorizado, controlado con placebo y entrecruzado, 42 sujetos sanos recibieron administraciones orales de dosis únicas de placebo, dolutegravir 250 mg en suspensión (exposiciones de aproximadamente 3 veces la dosis de 50 mg una vez al día en el estado estacionario) y moxifloxacino (400 mg, control activo) en secuencia aleatoria. Dolutegravir no prolongó el intervalo QTc durante 24 horas después de la dosis. Después del ajuste basal y para el placebo, la media máxima en el intervalo QTc con base en el método de corrección de Fridericia (QTcF) fue de 1.99 mseg (IC superior unilateral de 95%: 4.53 mseg). Efectos en la función renal: El efecto de dolutegravir en la depuración de creatinina sérica (CrCl), la tasa de filtración glomerular (GFR) utilizando iohexol como sustrato de prueba y el flujo plasmático renal eficaz (ERPF) utilizando para-aminohipurato (PAH) como sustrato de prueba, fue evaluado en un estudio abierto, aleatorizado, de 3 grupos, paralelo, controlado con placebo y realizado en 37 sujetos sanos que recibieron TIVICAY® 50 mg una vez al día (n=12), 50 mg dos veces al día (n=13) o placebo una vez al día (n=12) durante 14 días. Se observó una disminución modesta en la CrCl con dolutegravir dentro de la primera semana de tratamiento, la cual fue consistente con la observada en estudios clínicos. Dolutegravir administrado en ambas dosis no produjo efectos significativos en la GFR o ERPF. Estos datos respaldan los estudios in vitro que sugieren que los pequeños aumentos en las concentraciones de creatinina que se observaron en estudios clínicos se deben a la inhibición no patológica del transportador de cationes orgánicos 2 (OCT2) en los túbulos renales proximales, los cuales median la secreción tubular de creatinina. Estudios clínicos: Sujetos sin tratamiento previo a antirretrovirales: La eficacia de dolutegravir en sujetos infectados por VIH, vírgenes a tratamiento, se basa en los datos de dos estudios aleatorizados, internacionales, doble ciego, controlados con activo, datos de 96 semanas en SPRING-2 (ING113086) y SINGLE (ING114467). Esto es apoyado por datos de 48 semanas del estudio abierto controlado con activo FLAMINGO (ING114915). En SPRING, 822 adultos infectados con VIH-1, vírgenes a tratamiento antirretroviral (ART), fueron aleatorizados y recibieron al menos una dosis de TIVICAY® 50 mg una vez al día o raltegravir 400 mg dos veces al día, ambos administrados con un tratamiento dual con dosis fijas de NRTI (ABC/3TC o TDF/FTC). En la basal, la mediana de la edad de los pacientes fue de 36 años, 14% eran mujeres, 15% no blancos, y 12% tenían co infección por hepatitis B y/o C y 2% eran CDC Clase C; estas características eran similares entre grupos de tratamiento. En SINGLE, 833 sujetos fueron aleatorizados y recibieron al menos una dosis de TIVICAY® 50 mg una vez al día con una dosis fija de abacavir-lamivudina (TIVICAY® + ABC/3TC) o una dosis fija de efavirenz-tenofovir-emtricitabina (EFV/TDF/FTC). En la basal, la mediana de edad de los pacientes fue de 35 años, 16% eran mujeres, 32% no blancos, 7% tenían co infección por hepatitis C y 4% eran CDC Clase C, estas características eran similares entre grupos de tratamiento. Los objetivos primarios y otros resultados de la Semana 48 (incluyendo desenlaces por covariables basales clave) de SPRING-2 y SINGLE se muestran en la Tabla 2.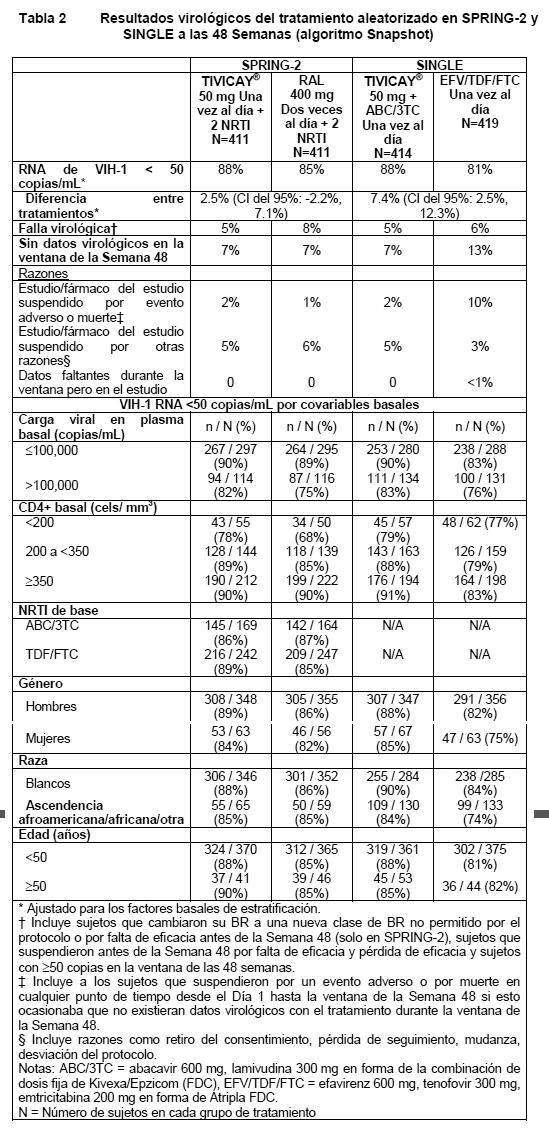
En el estudio SPRING-2, durante 96 semanas la supresión virológica (RNA de VIH-1 < 50 copias/mL) en el grupo de TIVICAY® (81%) no fue inferior al grupo de raltegravir (76%). La mediana de cambio en el conteo de células T CD4+ desde la basal fue de +230 cels/mm3 en el grupo que recibió TIVICAY y en el grupo de raltegravir a las 48 semanas y y 276 cel/mm3 en el grupo recibiendo dolutegravir comparado con 264 cels /mm3 del grupo de raltegravir a las 96 semanas. A la semana 48 del estudio SINGLE, la supresión virológica (RNA de VIH-1 < 50 copias/mL) en el grupo de TIVICAY® + ABC/3TC fue de 88%, la cual fue superior a la del grupo EFV/TDF/FTC (81%) en base al análisis primario (p= 0.003). A la semana 96 se mantenía la supresión virológica, el brazo TIVICAY® + ABC/3TC (80%) fue superior al brazo EFV/TDF/FTC (72%), la diferencia de los tratamientos fue 8.0 (2.3, 13.8), p=0.006. El cambio medio ajustado en el conteo de células T CD4+ desde la basal, fue de 267 cels/mm3 en el grupo que recibió TIVICAY® + ABC/3TC y 208 cels/mm3 en el grupo de EFV/TDF/FTC en SINGLE a las 48 semanas. La diferencia ajustada del CI del 95% fue de 58.9 (33.4, 84.4), p < 0.001 (modelo de mediciones repetidas ajustado para los factores de estratificación basales: RNA de VIH-1 basal y conteo basal de células T CD4+, entre otros factores). Este análisis fue pre especificado y ajustado para multiplicidad. La mediana de tiempo hasta la supresión viral fue de 28 días en el grupo que recibió TIVICAY® + ABC/3TC y de 84 días en el grupo de EFV/TDF/FTC en SINGLE a las 48 semanas (p < 0.0001). Este análisis fue pre especificado y ajustado para multiplicidad. Tanto en SPRING-2 como en SINGLE, las diferencias entre tratamientos para la supresión virológica (RNA de VIH-1 < 50 copias/mL) fueron comparables entre las características basales (género, raza y edad). Durante las 96 semanas en SINGLE, y SPRING-2, no se aislaron mutaciones de resistencia a INI ni de resistencia emergente con el tratamiento de base en los grupos que contenían dolutegravir. En SPRING-2, cuatro sujetos del grupo de raltegravir fallaron con mutaciones importantes de NRTI, y un sujeto desarrolló resistencia a raltegravir; en SINGLE, seis sujetos del grupo de EFV/TDF/FTC fallaron con mutaciones asociadas con resistencia a NNRTI y uno desarrolló una mutación importante de NRTI. En FLAMINGO (ING114915), un estudio abierto controlado con activo, 484 adultos infectados con VIH-1 que no habían recibido tratamiento antirretroviral fueron aleatorizados y recibieron una dosis ya sea de TIVICAY® 50 mg una vez al día o de darunavir/ritonavir (DRV/r) 800 mg/100 mg una vez al día, ambos administrados con NRTI dual de dosis fija (ABC/3TC o TDF/FTC). En la basal, la edad media de los pacientes fue de 34 años, 15% mujeres, 28% de raza no blanca, 10% tuvieron hepatitis B y/o co-infección C, y 3% fueron considerados Clase C de la CDC; estas características fueron similares entre los grupos de tratamiento. La supresión virológica (VIH-1 RNA < 50 copias/mL) en el grupo TIVICAY® (90%) fue superior a la del grupo DRV/r (83%) a la 48 semanas. La diferencia ajustada proporcional e IC 95% fue 7.1% (0.9, 13.2), p=0.025. Durante el tratamiento, no se observó la aparición de mutaciones de resistencia primaria al tratamiento con INI, PI o NRTI en los sujetos de los grupos de tratamiento TIVICAY® o DRV+RTV. Se demostró respuesta virológica sostenida en el estudio SPRING-1 (ING112276), en el cual 88% de los pacientes que recibieron TIVICAY® 50 mg (n= 51) una vez al día) tenían un RNA de VIH-1 < 50 copias/mL, en comparación con 72% de los pacientes del grupo de efavirenz (n= 50) a las 96 semanas. No se aislaron mutaciones de resistencia a INI ni de resistencia emergente con el tratamiento de base con TIVICAY® 50 mg una vez al día durante las 96 semanas. Sujetos con tratamiento antirretroviral previo (y vírgenes a inhibidores de integrasas): En el estudio SAILING internacional, multicéntrico, doble ciego (ING111762), 719 adultos infectados con VIH-1, con tratamiento previo con ART, fueron aleatorizados y recibieron TIVICAY® 50 mg una vez al día o raltegravir 400 mg dos veces al día con el régimen de base seleccionado por el investigador (BR), el cual consistió de hasta 2 agentes (incluyendo al menos un agente completamente activo). En la basal, la mediana de edad de los pacientes fue de 43 años, 32% eran mujeres, 50% no eran blancos, 16% tenían co infección por hepatitis B y/o C, y 46% eran CDC Clase C. Todos los sujetos tenían resistencia al menos a dos clases de ART, y 49% de los sujetos tenían resistencia al menos a 3 clases de ART en la basal. Los resultados de la Semana 48 (incluyendo los resultados por covariables basales clave) para SAILING se muestran en la Tabla 3.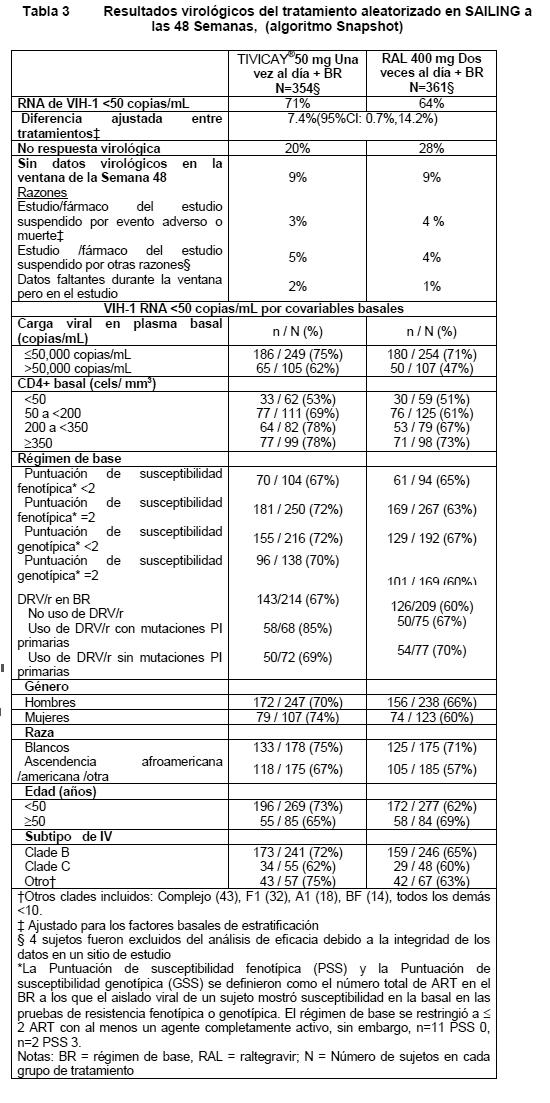
En el estudio SAILING, la supresión virológica (RNA de VIH-1 < 50 copias/mL) en el grupo de TIVICAY® (71%), fue estadísticamente superior a la del grupo de raltegravir (64%), a la Semana 48 (p= 0.030). Las diferencias entre tratamientos en cuanto a la supresión virológica (RNA de VIH-1 < 50 copias/mL) fueron comparables entre las características basales de género, raza, y subtipo de VIH. Los cambios medios en el conteo de células T CD4+ desde la basal, fueron de 113 cels/mm3 a la semana 24 y 162 cels/mm3 a la semana 48 en el grupo que recibió TIVICAY® y de 106 cels/mm3 a la semana 24 y 153 cels/mm3 en el grupo de raltegravir. Estadísticamente menos sujetos fallaron con el tratamiento con resistencia emergente con el tratamiento en el gen IN en el grupo de TIVICAY® (4/354, 0.1%) en comparación con raltegravir (17/361, 8.5%) (p= 0.003). Sujetos resistentes a inhibidores de integrasas: En el estudio piloto VIKING de Fase IIb, internacional, multicéntrico, abierto, de cohortes secuenciales de un solo grupo (ING112961), dos cohortes secuenciales de sujetos con resistencia a varias clases incluyendo resistencia a inhibidores de la integrasa de VIH, fueron reclutadas para examinar la actividad antiviral de TIVICAY® 50 mg una vez al día (n= 27) vs. 50 mg dos veces al día (n= 24) después de 10 días de monoterapia funcional. Las respuestas fueron mayores con la dosificación dos veces al día (1.8 log10 de cambio desde la basal en el RNA de VIH) en comparación con una vez al día (1.5 log10 de cambio desde la basal, diferencia ajustada de 0.3log10, p=0.017). Se mantuvieron tasas de respuesta más altas con la dosificación dos veces al día con dosis continuas de TIVICAY® y optimización del régimen de base durante las 48 semanas de tratamiento (33% vs. 71% < 50 c/mL, ITT-E, análisis TLOVR). Se observó un perfil de seguridad comparable entre dosis. Posteriormente, VIKING-3 examinó el efecto de TIVICAY® 50 mg dos veces al día durante 7 días de monoterapia funcional, seguido del tratamiento de base optimizado y tratamiento continuo con TIVICAY® dos veces al día. En el estudio multicéntrico, abierto, de un solo grupo, VIKING-3 (ING112574), se reclutaron adultos infectados con VIH-1, con tratamiento previo con ART, con falla virológica y evidencia concurrente o histórica de resistencia a raltegravir y/o elvitegravir, quienes recibieron TIVICAY® 50 mg dos veces al día con el régimen concurrente de base que había fallado durante 7 días, pero con ART de base optimizado desde el Día 8. Se reclutaron 183 sujetos, 133 con resistencia a INI en el Escrutinio, y 50 con solo evidencia histórica de resistencia (y no en el Escrutinio). En la basal, la mediana de edad de los pacientes era de 48 años, 23% eran mujeres, 29% no eran blancos, y 20% tenían co infección por hepatitis B y/o C. La mediana basal de CD4+ fue de 140 cels/mm3, la mediana de la duración del ART previo fue de 13 años, y 56% eran CDC Clase C. Los sujetos mostraron varias resistencias a distintas clases de ART en la basal: 79% tenían ≥2 mutaciones para NRTI, 75% ≥1 para NNRTI, y 71% ≥2 para PI; 62% tenían virus que no eran R5. El cambio medio desde la basal en el RNA de VIH el día 8 (criterio de valoración primario) fue de 1.4log10 (CI del 95% 1.3 - 1.5log10, p < 0.001). La respuesta se asoció con una mutación basal a INI, como se muestra en la Tabla 4.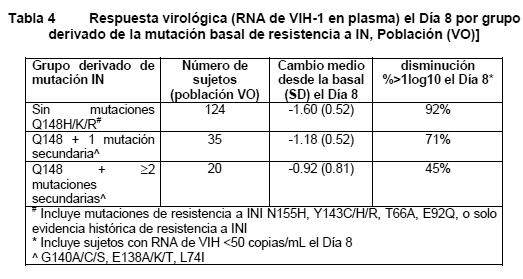
Después de la fase de monoterapia, los sujetos tuvieron la oportunidad de optimizar su régimen de base, siempre que fuera posible. De los 183 sujetos que terminaron las 24 semanas del estudio o que suspendieron antes del corte de datos, 126 (69%) tenían < 50 copias/mL de RNA en la Semana 24 (algoritmo Snapshot). Los sujetos que portaban virus con mutación Q148 con mutaciones secundarias adicionales asociadas con Q148, presentaron una respuesta más baja en la Semana 24. La puntuación de susceptibilidad general de base (OSS) no se asoció con respuesta en la Semana 24.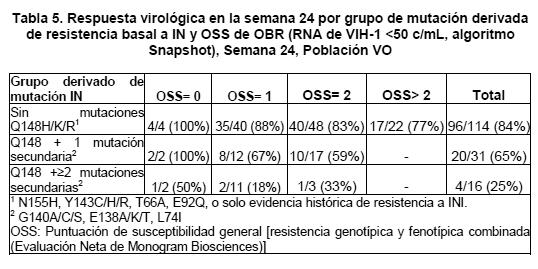
El índice de respuesta en la semana 48 fue sostenido, con 116/183 (63%) de los sujetos presentando VIH-1 RNA < 50 copias/mL (algoritmo "snapshot"). La respuesta también fue sostenida hasta la semana 48 en sujetos que portan el virus con mutación Q148 y con mutaciones secundarias relacionadas con Q148 adicionales. La proporción de sujetos con RNA-VIH < 50 copias/mL en la Semana 48 fue de 88/113 (78%) para mutaciones No Q148, 19/31 (61%) para Q148+1 y 4/16(25%) para Q148+≥2 mutaciones secundarias (población VO, algoritmo "snapshot"). La calificación de susceptibilidad general (OSS) de fondo no se relacionó con la respuesta de la Semana 48. La supresión virológica (RNA de VIH-1 < 50 copias/mL) fue comparable entre características basales (género, raza y edad). La mediana de cambio en el conteo de células T CD4+ desde la basal para VIKING-3 con base en datos observados fue de 61 cels/mm3 a la Semana 24 y 110 células/mm3 a la Semana 48 fue de 61 cels/mm3. En el estudio multicéntrico, doble ciego, controlado con placebo VIKING-4 (ING116529), 30 adultos infectados con VIH-1, previamente tratados con ART sin actual respuesta virológica en un régimen conteniendo un inhibidor de integrasa y resistencia genotípica primaria a INIs al escrutinio, fueron aleatorizados para recibir ya sea dolutegravir 50 mg dos veces al día o placebo con el actual régimen fallido durante 7 días con todos los sujetos recibiendo en forma abierta dolutegravir mas el régimen de base optimizado a partir del día 8. La comparación del objetivo primario de tratamiento al día 8, demostró que dolutegravir 50 mg dos veces al día fue superior al placebo, con una diferencia promedio ajustada del tratamiento en el cambio de los niveles basales en RNA de HIV-1 plasmático al día 8 de -1.2 log10 copias/mL (95% CI -1.5, -0.8 log10 copias/mL, p < 0.001). Las respuestas del día 8 en este estudio controlado con placebo fueron consistentes con las encontradas en VIKING-3, lo que incluye las categorías de resistencia basal a la integrasa. Niños: En un estudio multicéntrico, de Fase I/II de 48 semanas, abierto (P1093/ING112578), se evaluaron los parámetros farmacocinéticos, la seguridad, tolerabilidad y eficacia de TIVICAY® en regimenes combinados en lactantes, niños y adolescentes infectados con VIH-1. A las 24 semanas, 16 de 23 (70%) niños y adolescentes (12 a menos de 18 años de edad) tratados con TIVICAY® una vez al día (35 mg n= 4, 50 mg n= 19) más OBR, lograron una carga viral menor de 50 copias/mL. Veinte de 23 niños y adolescentes (87%) tuvieron una disminución > 1 log10 copias/mL a partir de la basal de RNA de VIH-1, ó de RNA de VIH-1 < 400 c/mL a la semana 24. Cuatro sujetos tuvieron resistencia virológica, ninguno de los cuales tenía resistencia a INI al momento de la resistencia virológica.
Contraindicaciones: TIVICAY® está contraindicado en combinación con dofetilide o pilsicainida. TIVICAY® está contraindicado en pacientes con hipersensibilidad conocida a dolutegravir o a cualquiera de los excipientes. No se use durante el embarazo, lactancia y pacientes menores de 12 años de edad.
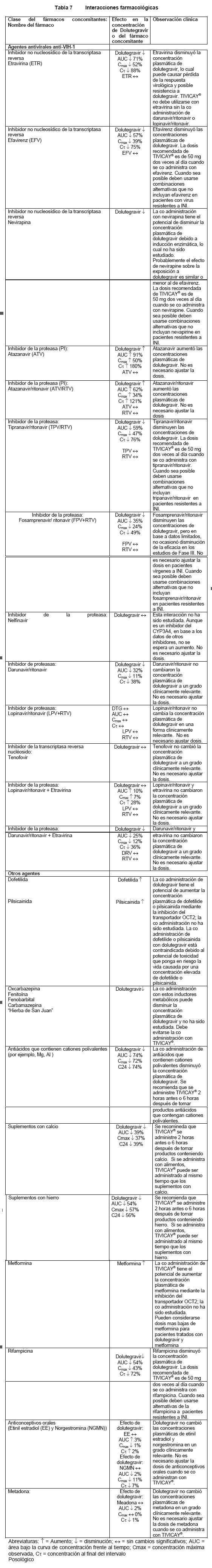
Precauciones generales: Reacciones de hipersensibilidad: Se han reportado reacciones de hipersensibilidad con inhibidores de integrasas, incluyendo TIVICAY®, las cuales se caracterizaron por erupción síntomas constitucionales, y algunas veces, disfunción orgánica incluyendo lesión hepática. Suspenda TIVICAY® y otros agentes sospechosos de inmediato si desarrolla signos o síntomas de reacciones de hipersensibilidad (incluyendo, sin limitarse a, erupción cutánea grave o erupción acompañada de fiebre, malestar general, fatiga, dolor muscular o articular, ampollas, úlceras orales, conjuntivitis, edema facial, hepatitis, eosinofilia, angioedema). Debe monitorearse el estado clínico, e incluir determinación de las transaminasas hepáticas, asi como iniciar el tratamiento adecuado.. El retraso en suspender el tratamiento con TIVICAY® u otros agentes etiológicos sospechosos después del inicio de la hipersensibilidad puede poner en riesgo la vida. Síndrome de Reconstitución Inmune: En pacientes infectados de VIH con severa inmunodeficiencia al momento de iniciar la terapia con antirretrovirales (ART), puede sobrevenir una reacción inflamatoria e infecciones oportunistas asintomáticas o residuales y causar una grave condición clínica, o exacerbación de los síntomas. Habitualmente, estas reacciones se observan en el transcurso de las primeras pocas semanas o meses de iniciar tratamiento con ART. Los ejemplos relevantes son retinitis por citomegalovirus, infecciones generalizadas y/o micobacterianas focales y neumonía por Pneumocystis jiroveci (P. carinii). Cualquier síntoma inflamatorio debe ser examinado sin dilación e iniciar el tratamiento cuando sea necesario. También se ha reportado que se presentan afecciones autoinmunes (como la enfermedad de Graves, polimiositis y el síndrome Guillain-Barré) al declararse la reconstitución inmune, sin embargo, el tiempo para declararse es más variable, y puede ocurrir muchos meses después de haber iniciado el tratamiento, y en ocasiones puede ser una presentación atípica. Se observaron aumentos de las pruebas de función hepática consistentes con el síndrome de reconstitución inmune en algunos pacientes co infectados con hepatitis B y/o C al momento de iniciar el tratamiento con TIVICAY®. Se recomienda el monitoreo de las pruebas de función hepática en pacientes con co infección por hepatitis B y/o C. Debe tenerse especial cuidado al iniciar o mantener un tratamiento eficaz contra la hepatitis B (relacionado con los lineamientos del tratamiento) al iniciar un tratamiento basado en dolutegravir en pacientes co infectados por hepatitis B (ver Reacciones adversas). Infecciones oportunistas: Los pacientes a los que se les administra TIVICAY® o cualquier otra terapia antirretroviral todavía pueden desarrollar infecciones oportunistas y otras complicaciones por la infección de VIH. Por ende los pacientes deben permanecer bajo cuidadosa observación clínica en manos de médicos experimentados en el tratamiento de estas enfermedades asociadas al VIH. Transmisión de la infección: A los pacientes se les debe informar que no se ha demostrado que la terapia antirretroviral actual, incluyendo a TIVICAY®, pueda evitar el riesgo de transmisión de VIH a otras personas, por medio de una relación sexual o de sangre contaminada. Deben seguir tomando las debidas precauciones. Interacción entre Fármacos: Deben tomarse precauciones al coadministrar medicamentos (recetados o sin receta) que pueden cambiar la exposición de TIVICAY® o medicamentos en los que TIVICAY® pueda cambiar su exposición (vea Contraindicaciones e Interacciones). La coadministración de TIVICAY® con etravirina (ETR) sólo se permite si el paciente también está recibiendo simultáneamente atazanavir+ritonavir (ATV/RTV), lopinavir+ritonavir (LPV/RTV) o darunavir + ritonavir (DRV/RTV) (vea Interacciones). La dosis recomendada de TIVICAY® es de 50 mg dos veces al día cuando se co administra con efavirenz, nevirapina, tipranavir/ritonavir, o rifampicina (ver Interacciones). TIVICAY® no debe coadministrarse con antiácidos que contengan cationes polivalentes. Estos agentes deben administrarse 2 horas después o 6 horas antes de TIVICAY® (vea Interacciones). Se recomienda administrar TIVICAY® dos horas antes ó seis horas después de tomar suplementos de calcio o hierro, o alternativamente, administrarse con alimentos (ver Interacciones). Las concentraciones de metformina pueden incrementarse por TIVICAY®. Debe monitorearse a los sujetos durante la terapia y puede requerirse un ajuste de dosis de metformina (vea Interacciones).
Efectos sobre la capacidad de conducir y operar maquinaria: No se han realizado estudios para investigar el efecto de TIVICAY® sobre el desempeño al conducir vehículos o la capacidad para utilizar maquinaria. Debe tenerse en cuenta el estado clínico del paciente y el perfil de eventos adversos de TIVICAY® al considerar la capacidad del paciente para manejar o utilizar maquinaria.
Restricciones de uso durante el embarazo y la lactancia: Fertilidad: No existen datos acerca de los efectos de TIVICAY® sobre la fertilidad humana masculina o femenina. Los estudios realizados en animales no indican que dolutegravir produzca efectos en la fertilidad masculina o femenina (véase Información no clínica). Embarazo: No se han realizado estudios adecuados y bien controlados de TIVICAY® en mujeres embarazadas