TOMZY
ZYDUS
Denominación genérica: Topiramato.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: Topiramato 25 mg, 50 mg y 100 mg. Excipiente, c.b.p. 1 tableta.
Indicaciones terapéuticas: Epilepsia: Topiramato está indicado como monoterapia en pacientes con epilepsia de reciente diagnóstico o también para la conversión a monoterapia en pacientes con epilepsia. Topiramato está indicado como terapia adjunta en adultos y niños (mayores de 2 años) con crisis de inicio parcial y crisis generalizadas tónico-clónicas. Topiramato también está indicado en adultos y niños como terapia adjunta en el tratamiento de convulsiones asociadas con el síndrome de Lennox-Gastaut. Migraña: Topiramato está indicado en adultos para el tratamiento profiláctico de la cefalea migrañosa. La utilidad de topiramato en el tratamiento agudo de cefaleas migrañosas no ha sido estudiada.
Farmacocinética y farmacodinamia: Farmacocinética: El perfil farmacocinético de topiramato en comparación con otros fármacos antiepilépticos muestra una vida media plasmática larga, farmacocinética lineal, depuración predominantemente renal, ausencia de unión significativa a proteínas y falta de metabolitos activos clínicamente relevantes. El topiramato no es un potente inductor de enzimas metabolizadoras de fármacos, puede ser administrado sin considerar los alimentos, y el monitoreo rutinario de concentraciones plasmáticas no es necesario. En estudios clínicos, no hubo una relación consistente entre las concentraciones plasmáticas y la eficacia o eventos adversos. El topiramato se absorbe bien y de manera rápida. Después de la administración oral de 100 mg de topiramato a sujetos sanos, el pico de la concentración plasmática media (Cmáx) de 1.5 mg/ml se alcanzó dentro de 2 a 3 horas (Tmáx). En base a la recuperación de la radiactividad de la orina, el grado medio de absorción de una dosis oral de 100 mg de 14C-topiramato fue al menos de 81%. No existe efecto clínicamente significativo de la comida sobre la biodisponibilidad de topiramato. Generalmente 13-17% de topiramato está unido a las proteínas del plasma. Se ha observado una baja capacidad de unión en sitio para topiramato en/sobre eritrocitos que es saturable por debajo de concentraciones plasmáticas de 4 mg/ml. El volumen de distribución varió inversamente con la dosis. El volumen aparente medio de distribución fue 0.8 a 0.55 L/kg para dosis sencillas en un rango de 100 hasta 1,200 mg. Se detectó un efecto del género sobre el volumen de distribución, siendo los valores para mujeres cerca de 50% de aquellos de los hombres. Esto se atribuye al mayor porcentaje de grasa corporal en mujeres y no es de consecuencia significativa. El topiramato no se metaboliza extensamente (~ 20%) en voluntarios sanos. Topiramato se metaboliza hasta 50% en pacientes que reciben terapia antiepiléptica paralela con inductores conocidos de enzimas metabolizadoras de fármacos. Se han aislado seis metabolitos formados a partir de hidroxilación, hidrólisis y glucuronidación, caracterizados e identificados a partir de plasma, orina y heces en humanos. Cada metabolito representa menos de 3% de la radiactividad total excretada después de la administración de 14C-topiramato. Dos metabolitos, que fueron retenidos en la mayoría de la estructura del topiramato fueron analizados y se encontró que tenía una pequeña actividad anticonvulsiva o bien, no tenían ninguna actividad. En humanos, la principal vía de eliminación de topiramato sin cambios y sus metabolitos es el riñón (por lo menos 81% de la dosis). Aproximadamente 66% de una dosis de 14C-topiramato fue excretada sin cambios en la orina dentro de cuatro días. Después de una dosis de 50 y 100 mg de topiramato dos veces al día, la depuración renal media fue de aproximadamente 18 y 17 ml/min respectivamente. Existe evidencia de reabsorción tubular renal de topiramato. Esto es soportado por estudios en ratas donde el topiramato fue coadministrado con probenecid, y se observó un aumento significativo en la depuración renal de topiramato. En general, la depuración plasmática es aproximadamente de 20 a 30 ml/min en humanos después de la administración oral. El topiramato presenta baja variabilidad inter-sujeto en las concentraciones plasmáticas y, por lo tanto, su farmacocinética es predecible. La farmacocinética de topiramato es lineal con la depuración del plasma permaneciendo constante, y el área bajo la curva de la concentración en plasma aumentando de manera proporcional a la dosis en un rango de 100 a 400 mg de dosis individuales en sujetos sanos. Los pacientes con función renal normal pueden tardar 4 a 8 días en alcanzar concentraciones de plasma de estado estable. La Cmáx media, después de dosis orales múltiples de 100 mg dos veces al día a sujetos sanos fue de 6.76 m/ml. Después de una administración de dosis múltiples de 50 a 100 mg de topiramato dos veces al día, la vida media de la eliminación plasmática fue de aproximadamente 21 horas. La administración concomitante de dosis múltiples de topiramato, 100 a 400 mg dos veces al día, con fenitoína o carbamazepina muestra aumentos proporcionales a la dosis en concentraciones plasmáticas de topiramato. La depuración plasmática y renal de topiramato disminuye en pacientes con función renal deteriorada (ClCR < 60 ml/min) y la depuración plasmática disminuye en pacientes con padecimiento renal en etapa terminal. Como resultado, se esperan concentraciones plasmáticas de topiramato en estado estable más altas para una dosis dada en pacientes con deterioro renal en comparación con aquéllos con una función renal normal. El topiramato es removido efectivamente del plasma por hemodiálisis. La depuración plasmática de topiramato disminuye en pacientes con deterioro hepático moderado a severo. La depuración plasmática de topiramato no cambia en sujetos de edad madura, en ausencia de padecimiento renal. Farmacocinética pediátrica hasta los 12 años de edad: La farmacocinética de topiramato en niños, al igual que en los adultos que reciben terapia adyuvante es lineal con una depuración independiente de la dosis y de las concentraciones plasmáticas en estado estable aumentando en proporción a la dosis. Los niños, sin embargo, tienen una depuración más alta y una vida media de eliminación más corta. En consecuencia, las concentraciones plasmáticas de topiramato para la misma dosis mg/kg puede ser menor en niños en comparación con adultos. Al igual que en adultos, las enzimas hepáticas inducidas por fármacos antiepilépticos disminuyen las concentraciones plasmáticas en estado estable.
Contraindicaciones: Hipersensibilidad a cualquiera de los componentes de la fórmula.
Precauciones generales: En pacientes con o sin historia de convulsiones o epilepsia, los fármacos antiepilépticos, incluyendo topiramato, deben retirarse gradualmente para minimizar el potencial de convulsiones o incrementar la frecuencia del ataque. En estudios clínicos, las dosis diarias se redujeron en intervalos semanales de 50-100 mg en adultos con epilepsia y de 25-50 mg en adultos que recibieron topiramato a dosis arriba de 100 mg/día en profilaxis de migraña. En estudios clínicos en niños, topiramato se retiró gradualmente en un intervalo de 2-8 semanas. En situaciones en las que un rápido retiro de topiramato es médicamente requerido, se recomienda un monitoreo apropiado. La principal vía de eliminación de topiramato inalterado y sus metabolitos es el riñón. La eliminación renal es dependiente de la función renal y es independiente de la edad. Los pacientes con deterioro renal moderado o severo pueden requerir 10 a 15 días para alcanzar las concentraciones plasmáticas en estado de equilibrio, comparado con 4 a 8 días en pacientes con función renal normal. Al igual que todos los pacientes, el programa de titulación se debe guiar por los resultados clínicos (por ejemplo, control de crisis, evitando efectos adversos) con el conocimiento de que los pacientes con deterioro renal conocido pueden requerir un mayor tiempo para alcanzar el estado de equilibrio en cada dosis. Es muy importante la hidratación adecuada mientras se toma topiramato. La hidratación puede reducir el riesgo de nefrolitiasis. La hidratación adecuada antes y durante las actividades como ejercicio o exposición a temperaturas altas puede reducir el riesgo de eventos adversos relativos al calor. Alteraciones del estado de ánimo/depresión: Se ha observado un aumento en la incidencia de alteraciones del estado de ánimo y depresión durante el tratamiento con topiramato. Intento de suicidio: En las fases doble ciego de estudios clínicos con topiramato en indicaciones aprobadas y de investigación, ocurrieron intentos de suicidio en una tasa de 0.003 (13 eventos/3,999 pacientes años) con topiramato contra 0 (0 eventos/1,430 pacientes años) con placebo. Se reportó un suicidio en un paciente que recibía topiramato dentro de un estudio de desorden bipolar. Por esta razón los pacientes deben ser monitorizados para identificar signos de ideación o comportamiento suicida y considerar el tratamiento apropiado. Debe recomendarse a los pacientes y cuidadores sobre la necesidad de evaluación médica en caso de observar signos de comportamiento o ideación suicida. Nefrolitiasis: Algunos pacientes, especialmente aquéllos con predisposición a la nefrolitiasis, pueden estar en riesgo de formación de cálculos renales y signos y síntomas asociados como cólicos renales, dolor renal o dolor en el costado. Los factores de riesgo para nefrolitiasis incluyen formación de cálculos previos, historia familiar de nefrolitiasis e hipercalciuria. Ninguno de estos riesgos puede realmente predecir la formación de cálculos durante el tratamiento con topiramato. Además, los pacientes que están tomando otros medicamentos asociados con nefrolitiasis pueden incrementar el riesgo. Función hepática disminuida: En pacientes con deterioro hepático, el topiramato se debe administrar con precaución ya que la depuración de topiramato puede disminuir. Miopía aguda y glaucoma secundario de ángulo cerrado: Se ha reportado un síndrome que consiste en miopía aguda con glaucoma secundario de ángulo cerrado en pacientes que recibieron topiramato. Los síntomas incluyen el inicio agudo de agudeza visual disminuida y/o dolor ocular. Los hallazgos oftalmológicos pueden incluir miopía, cámara anterior superficial, hiperemia ocular (enrojecimiento) y aumento en la presión intraocular. Puede o no haber midriasis. Este síndrome puede estar asociado con efusión supraciliar resultando en desplazamiento anterior de la lente e iris con glaucoma secundario de ángulo cerrado. Los síntomas ocurren típicamente dentro de 1 mes del inicio de la terapia con topiramato. En contraste con el glaucoma primario de ángulo estrecho, que es raro en personas menores de 40 años de edad, el glaucoma secundario de ángulo cerrado asociado con topiramato se ha reportado en pacientes pediátricos y en adultos. El tratamiento incluye descontinuación de topiramato tan rápido como sea posible a juicio del médico tratante, y medidas apropiadas para reducir la presión intraocular. Estas medidas generalmente resultan en una disminución de la presión intraocular (la presión intraocular elevada de cualquier etiología, si no es tratada, puede producir secuelas serias incluyendo pérdida permanente de la visión). Acidosis metabólica: La acidosis metabólica, hiperclorémica, de brecha no aniónica (esto es bicarbonato sérico disminuido por debajo del rango de referencia normal en la ausencia de alcalosis respiratoria) está asociada con el tratamiento con topiramato. Esta disminución en el bicarbonato sérico es debido al efecto inhibitorio de topiramato sobre la anhidrasa carbónica renal. Generalmente, la disminución en bicarbonato ocurre temprano en el tratamiento aunque puede ocurrir en cualquier momento durante el tratamiento. Estas disminuciones son normalmente leves a moderadas (disminución promedio de 4 mmol/L a dosis de 100 mg/día o mayor en adultos y aproximadamente 6 mg/kg/día en niños. Raramente, los pacientes han experimentado disminuciones a valores por debajo de 10 mmol/L. Las condiciones o terapias que predisponen a acidosis (como enfermedad renal, alteraciones respiratorias severas, status epilepticus, diarrea, cirugía, dieta cetogénica, o ciertos fármacos) pueden ser aditivos a los efectos del topiramato de disminución del bicarbonato. La acidosis metabólica crónica en pacientes pediátricos puede reducir las tasas de crecimiento. El efecto de topiramato sobre el crecimiento y las secuelas relacionadas con los huesos no han sido sistemáticamente investigadas en poblaciones pediátricas o de adultos. Dependiendo de las condiciones subyacentes, con la terapia con topiramato se recomienda la evaluación apropiada incluyendo los niveles séricos de bicarbonato. Si la acidosis metabólica se desarrolla y persiste, se deberán tener consideraciones para reducir la dosis o descontinuar el topiramato (utilizando disminución de la dosis). Suplementación nutricional: Se puede considerar una dieta suplementaria o aumento en la ingesta de alimentos si el paciente está perdiendo peso mientras toma este medicamento.
Restricciones de uso durante el embarazo y la lactancia: No existen estudios controlados en personas embarazadas o uso de topiramato, sin embargo la información de registro sugiere que podría haber una asociación entre el uso de topiramato durante el embarazo y malformaciones congénitas (defectos craneofaciales como labio y paladar hendido; además de hipospadias y anormalidades que involucra varios sistemas corporales). Adicionalmente podría haber un riesgo incrementado de efectos teratogénicos asociados con el uso de medicamentos antiepilépticos en la terapia combinada. La paciente deberá ser comunicada del riesgo potencial del feto. Topiramato deberá ser utilizado durante el embarazo sólo si el potencial beneficio sobrepasa el riego potencial. En estudios preclínicos, topiramato ha mostrado tener efectos teratogénicos en las especies estudiadas (ratones, ratas y conejos). En ratas, el topiramato cruza la barrera placentaria. Topiramato se excretó en la leche de ratas lactando. La excreción de topiramato en la leche humana no ha sido evaluada en estudios controlados. Las observaciones limitadas en pacientes sugieren una amplia excreción de topiramato en la leche materna. Debido a que muchos fármacos se excretan en la leche humana, se debe tomar la decisión de descontinuar la lactancia o el fármaco, tomando en cuenta la importancia del fármaco para la madre. En la experiencia post-comercialización, se han reportado casos de hipospadias en infantes varones expuestos in utero a topiramato, con o sin otros anticonvulsivantes; sin embargo, no se ha establecido una relación causal con topiramato. No se use en el embarazo y la lactancia.
Reacciones secundarias y adversas: La mayoría de los eventos adversos más comunes en estudios clínicos fueron de severidad leve a moderada y relacionada con la dosis. Esos eventos adversos relacionados con la dosis empezaron típicamente en la fase de la titulación y a menudo persistieron en la fase de mantenimiento pero empezaron pocas veces en la fase de mantenimiento. Una velocidad rápida de titulación y dosis iniciales más altas se asociaron con incidencias más altas de eventos adversos que resultaron en la discontinuación. Ya que topiramato ha sido más frecuentemente coadministrado con otros agentes antiepilépticos, no es posible determinar qué agentes, si los hay, están asociados con eventos adversos. En estudios clínicos doble ciego, controlados con placebo, algunos de los cuales incluyeron un periodo rápido de titulación, los eventos adversos ocurrieron con una frecuencia mayor o igual a 5%, y con una mayor incidencia en los pacientes adultos tratados con topiramato que en el grupo placebo e incluyeron: somnolencia, vértigo, nerviosismo, ansiedad, ataxia, fatiga, desórdenes del habla, lentitud psicomotora, visión anormal, dificultad con la memoria NOS*, confusión, parestesia, diplopía, anorexia, nistagmo, náusea, diarrea, dispepsia, boca seca, alteraciones del gusto, hipoestesia, insomnio, disminución de peso, problemas de lenguaje, dificultad con concentración/atención, depresión, dolor abdominal, astenia y alteraciones en el estado de ánimo. En niños también la marcha anormal y reacciones agresivas, alteraciones de personalidad e hiperquinesia. Los eventos adversos menos frecuentes pero considerados potencialmente médicamente relevantes incluyeron: alteraciones del gusto, agitación, problemas cognoscitivos NOS*, labilidad emocional, problemas de coordinación, marcha anormal, apatía, psicosis/síntomas psicóticos, comportamiento/reacciones agresivas, leucopenia y nefrolitiasis. También se han reportado casos aislados de eventos tromboembólicos; sin embargo, no se estableció relación causal con el medicamento. En niños también se detectaron: Lentitud psicomotora, confusión, alucinaciones y depresión. El tratamiento en niños de dos años de edad o menos debe iniciarse a dosis de 1 mg/kg por las noches durante la primer semana. La dosis debe incrementarse a intervalos de una o dos semanas a razón de 0.5-1 mg/kg/día, dividido en dos dosis al día. Si el niño no tolera este régimen de titulación, pueden hacerse menores incrementos o intervalos mayores entre cada incremento de la dosis. La dosis y dosis de titulación deben ser dirigidas por la respuesta clínica. El rango recomendado de la dosis de titulación inicial para la monoterapia con topiramato en niños de dos años o más es de 100 a 400 mg/día o 3-6 mg/kg/día. Niños con diagnóstico reciente de crisis parciales han recibido dosis mayores a 500 mg/día. Experiencia poscomercialización y de otro tipo: Las reacciones adversas del fármaco de reportes espontáneos a partir de la comercialización mundial con topiramato se incluyen en la tabla siguiente. Las reacciones adversas del fármaco se clasificaron por frecuencia de la siguiente manera: Muy común: ≥ 1/10. Común: ≥ 1/100 y < 1/10. Poco común: ≥ 1/1,000 y < 1/100. Raro: ≥ 1/10,000 y < 1/1,000. Muy raro: ≥ 1/10,000. Reportes post-comercialización de reacciones adversas del fármaco: Alteraciones de la sangre y el sistema linfático: Muy raro: leucopenia, neutropenia y trombocitopenia. Raro: anorexia. Alteraciones del metabolismo y nutrición: Muy raro: acidosis metabólica (véase Precauciones generales); disminución del apetito, hiperamonemia (véase Interacciones medicamentosas y de otro género). Alteraciones psiquiátricas: Raro: depresión (véase Precauciones generales); agitación; somnolencia. Muy raro: insomnio, estado de confusión, trastorno psicótico, agresión, alucinaciones, ideas suicidas, intentos y suicidio (véase Precauciones generales); desorden del lenguaje expresivo. Alteraciones del sistema nervioso: Raro: parestesia, convulsión, cefalea. Muy raro: desorden del habla, disgeusia, amnesia, deterioro de la memoria. Alteraciones del ojo: Raro: trastornos visuales. Muy raro: miopía, glaucoma de ángulo cerrado (véase Precauciones generales); dolor de ojo. Muy raro: maculopatía. Alteraciones gastrointestinales: Raro: náusea. Muy raro: diarrea, dolor abdominal, vómito. Alteraciones de la piel y tejido subcutáneo: Raro: alopecia. Muy raro: erupción. Alteraciones renales y urinarios: Raro: nefrolitiasis (véase Precauciones generales). Alteraciones generales y condiciones en el sitio de administración: Raro: fatiga. Muy raro: pirexia, sentimiento anormal, astenia. Investigaciones: Raro: disminución de peso. Se han recibido reportes aislados sobre hepatitis e insuficiencia hepática en pacientes que tomaron múltiples medicamentos, incluyendo topiramato. También se han recibido reportes aislados de ámpulas en la piel y reacciones de las mucosas (incluyendo eritema multiforme. pénfigos, síndrome de Stevens-Johnson, y necrólisis epidérmica tóxica); la mayoría de estos reportes han ocurrido en pacientes que tomaron otros medicamentos también asociados con estas lesiones de piel y mucosas. Se ha reportado raramente oligohidrosis con el uso de topiramato, siendo la mayoría de estos reportes en niños.
Interacciones medicamentosas y de otro género: (Para propósitos de esta sección, una dosis sin efecto se define como un cambio ? 15%). Efectos de topiramato sobre otros fármacos antiepilépticos: La adición de topiramato a otros medicamentos antiepilépticos (fenitoína, carbamazepina, ácido valproico, fenobarbital, primidona) no tiene efecto en sus concentraciones plasmáticas de estado estable, excepto en el paciente ocasional, donde la adición de topiramato a la fenitoína puede conducir a un aumento en las concentraciones plasmáticas de fenitoína. Esto es posiblemente debido a la inhibición de una isoforma polimórfica de enzima específica (CYP2C19). En consecuencia, en cualquier paciente bajo tratamiento con fenitoína que muestre signos o síntomas de toxicidad, deben monitorearse los niveles de fenitoína. Un estudio de interacción farmacocinética de pacientes con epilepsia indicó que la adición de topiramato a lamotrigina no tuvo efecto sobre la concentración plasmática de estado estable de lamotrigina a dosis de topiramato de 100 a 400 mg/día. Además, no hubo cambios en la concentración plasmática en estado estable de topiramato durante o después del retiro del tratamiento con lamotrigina (dosis media de 327 mg/día). Efectos de otros fármacos antiepilépticos sobre topiramato: La fenitoína y la carbamazepina disminuyen las concentraciones plasmáticas de topiramato. La adición o el retiro de fenitoína o carbamazepina a la terapia con topiramato, puede requerir un ajuste en la dosis de este último. Esto debe realizarse mediante seguimiento del efecto clínico. La adición o el retiro de ácido valpróico no produce cambios clínicamente significativos en las concentraciones de topiramato en plasma y, por lo tanto, no justifica un ajuste en la dosificación de topiramato. Los resultados de estas interacciones se resumen a continuación: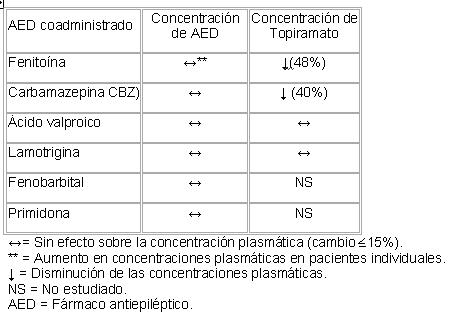
Otras interacciones medicamentosas: Digoxina: En un estudio con dosis sencillas, el área bajo la curva de concentración plasmática de digoxina sérica (ABC) disminuyó 12% debido a la administración paralela con topiramato. La relevancia clínica a esta observación aún no ha sido establecida. Cuando se adiciona o retira topiramato en pacientes bajo terapia con digoxina debe prestarse atención particular al monitoreo de rutina de digoxina en suero. Depresores del SNC: La administración simultánea de topiramato y alcohol u otros medicamentos depresores del SNC no ha sido evaluada en estudios clínicos. Se recomienda no emplear topiramato paralelamente con alcohol y otras drogas depresoras del SNC. Anticonceptivos orales: En un estudio de interacción farmacocinética en voluntarios sanos con administración concomitante de anticonceptivos orales de combinación conteniendo 1 mg de noretindrona (NET) más 35 mg de etinilestradiol (EE), topiramato administrado en ausencia de otros medicamentos a dosis de 50 a 200 mg/día no estuvo asociado con cambios estadísticamente significativos en la exposición media (ABC) a cualquier componente del contraceptivo oral. En otro estudio, la exposición a EE fue disminuida estadísticamente significante a dosis de 200, 400 y 800 mg/día (18, 21 y 30%, respectivamente) cuando se administró como terapia adjunta en pacientes que tomaron ácido valpróico. En ambos estudios topiramato (50 mg/día a 800 mg/día) no afectó significativamente la exposición a NET. Aunque hubo una disminución dependiente de la dosis en exposición de EE para dosis entre 200-800 mg/día, no hubo un cambio significativo dependiente de la dosis en exposición de EE para dosis de 50-200 mg/día. La significancia clínica de los cambios observados se desconoce. Se debe considerar la posibilidad de una disminución de eficacia contraceptiva y aumento en sangrado irruptivo en pacientes que toman productos contraceptivos orales de combinación con topiramato. Se deberá solicitar a las pacientes que toman contraceptivos que contienen estrógenos, que reporten cualquier cambio en sus patrones de sangrado. La eficacia contraceptiva puede verse disminuida aun en la ausencia de sangrado irruptivo. Litio: En voluntarios sanos, se observó una reducción (18% para ABC) en exposición sistémica para litio durante la administración concomitante con topiramato 200 mg/día. En pacientes con desorden bipolar, la farmacocinética de litio no se vio efectada durante el tratamiento con topiramato a dosis de 200 mg/día; sin embargo, hubo un aumento observado en exposición sistémica (26% para ABC) después de dosis de topiramato de hasta 600 mg/día. Los niveles de litio deberán ser monitoreados cuando se coadministran con topiramato. Risperidona: Los estudios de interacción fármaco-fármaco conducidos bajo condiciones de dosis individual y múltiple en voluntarios sanos y en pacientes con desorden bipolar produjeron resultados similares. Cuando se administró concomitantemente con topiramato a dosis de escalamiento de 100, 1,250 y 400 mg/día hubo una reducción en la exposición sistémica (16 y 33% para ABC de estado estable en las dosis de 250 y 400 mg/día, respectivamente) de risperidona (administrada como dosis de 1 a 6 mg/día). Se observaron alteraciones mínimas en la farmacocinética de la mitad activa total (risperidona más 9-hidroxirisperidona) y sin alteraciones para 9-hidroxirisperidona. No hubo cambios clínicamente significativos en la exposición sistémica de la mitad activa total de risperidona o de topiramato, por lo tanto, no es probable que esta interacción sea de significancia clínica. Hidroclorotiazida (HCTZ): Un estudio de interacción fármaco-fármaco conducido en voluntarios sanos evaluó la farmacocinética de estado-estable de HCTZ (25 mg c/24 horas) y topiramato (96 mg c/12 horas) cuando se administraron solas y concomitantemente. Los resultados de este estudio indicaron que la Cmáx. de topiramato aumentó 27% y el ABC aumentó 29% cuando se añadió HCTZ a topiramato. La significancia clínica de este cambio se desconoce. La adición de HCTZ a la terapia con topiramato puede requerir un ajuste de la dosis de topiramato. La farmacocinética de HCTZ en estado estable no se vió influenciada significativamente por la administración concomitante de topiramato. Los resultados de laboratorio clínico indicaron disminuciones en el potasio sérico después de la administración de topiramato o HCTZ, que fueron mayores cuando HCTZ y topiramato fueron administrados en combinación. Metformina: Un estudio de interacción fármaco-fármaco conducido en voluntarios sanos evaluó la farmacocinética en estado estable de metformina y topiramato en plasma cuando se administró metformina sola y cuando se administraron simultáneamente metformina y topiramato. Los resultados de este estudio indicaron que la Cmáx. y ABC0-12h media aumentaron 18 y 25% respectivamente, mientras que la CL/F media disminuyó 20% cuando la metformina fue coadministrada con topiramato. El topiramato no afectó el Tmáx. de metformina. La significancia clínica del efecto de topiramato sobre la farmacocinética de metformina no es clara. La depuración plasmática oral de topiramato parece estar reducida cuando se administra con metformina. El grado de cambio en la depuración se desconoce. La significancia clínica del efecto de metformina sobre la farmacocinética de topiramato no es clara. Cuando topiramato se añade o retira en pacientes en terapia con metformina, se debe prestar especial atención al monitoreo de rutina para el control adecuado de su estado diabético. Pioglitazona: Un estudio de interacción fármaco-fármaco conducido en voluntarios sanos evaluó la farmacocinética en estado estable de topiramato y pioglitazona cuando se administraron solos y concomitantemente. Se observó una disminución de 15% en el ABCt, SS de pioglitazona sin alteración en Cmáx, SS. Este hallazgo no fue estadísticamente significativo. Además, se advirtió una disminución de 13 y 16% en Cmáx, SS y ABCt, SS respectivamente, del metabolito hidroxi activo así como una disminución de 60% en Cmáx, SS y ABCt, SS del metabolito ceto-activo. La significancia clínica de estos hallazgos se desconoce. Cuando topiramato se añade a la terapia con pioglitazona o la pioglitazona se añade a la terapia con topiramato, se debe prestar atención cuidadosa al monitoreo de rutina de pacientes para el control adecuado de su estado de enfermedad diabética. Gliburida: Un estudio de interacción fármaco-fármaco conducido en pacientes con diabetes tipo 2 evaluó la farmacocinética de estado estable de gliburida (5 mg/día) solo y concomitantemente con topiramato (150 mg/día). Hubo una reducción de 25% en ABC24 de gliburida durante la administración con topiramato. La exposición sistémica de los metabolitos activos, 4-trans-hidroxi-gliburida (M1) y 3-cis-hidroxigliburida (M2), también se redujeron 13 y 15% respectivamente. La farmacocinética de estado estable de topiramato no se vio afectada por la administración concomitante de gliburida. Cuando se añade topiramato a la terapia con gliburida o cuando la gliburida se añade a la terapia con topiramato, se debe prestar atención cuidadosa al monitoreo de rutina de pacientes para el control adecuado de su enfermedad diabética. Otras formas de interacción: Agentes de predisposición a nefrolitiasis: Topiramato, utilizado concomitantemente con otros agentes de predisposición a la nefrolitiasis, puede aumentar el riesgo de nefrolitiasis. Cuando se utiliza topiramato se debe evitar agentes como estos ya que pueden crear un ambiente fisiológico que aumente el riesgo de la formación de cálculos renales. Ácido valpróico: La administración concomitante de topiramato y ácido valpróico se ha asociado con hiperamonemia con o sin encefalopatía en pacientes que han tolerado cualquier fármaco solo. En la mayoría de los casos, los signos y síntomas disminuyeron con la descontinuación de cualquier fármaco. Este evento adverso no es debido a una interacción farmacocinética. No se ha establecido una asociación de hiperamonemia con la monoterapia de topiramato o tratamiento concomitante con otros antiepilépticos. Estudios farmacocinéticos de interacción de fármacos adicionales: Se han conducido estudios clínicos para evaluar el potencial de interacción farmacocinética de fármacos entre topiramato y otros agentes. A continuación se resumen los cambios en Cmáx o ABC como un resultado de las interacciones. La segunda columna (concentración de fármaco concomitante) describe qué pasa a la concentración del fármaco concomitante listado en la primera columna cuando se añade topiramato. La tercera columna (concentración de topiramato) describe como la coadministración de un fármaco listado en la primera columna modifica la concentración de topiramato. Resumen de resultados de estudios adicionales de farmacocinética clínica de interacción de fármacos: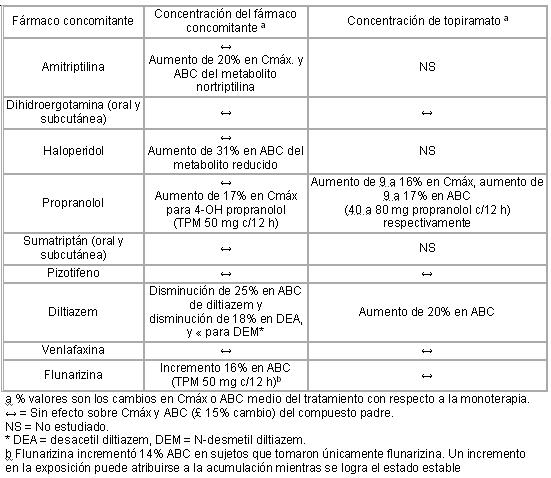
Alteraciones en los resultados de pruebas de laboratorio: Los datos de estudios clínicos indican que topiramato se ha asociado con una disminución promedio de 4 mmol/L en el nivel de bicarbonato en suero.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: La exposición aguda, y a largo plazo de ratones, ratas, perros y conejos a topiramato fue bien tolerada. Se observó hiperplasia de las células epiteliales gástricas sólo en roedores y en ratas, fue reversible después de 9 semanas sin tratamiento. Se observaron tumores de músculo liso en la vejiga urinaria sólo en ratones (dosis orales de hasta 300 mg/kg durante 21 meses) y pareció ser la única de las especies. Debido a que no existe una contraparte humana, estos no fueron considerados clínicamente relevantes. No ocurrieron estos hallazgos en el estudio de carcinogenicidad en ratas (dosis orales hasta 120 mg/kg/día durante 24 meses). Otros efectos toxicológicos y patológicos de topiramato observados en estos estudios pueden estar relacionados con la débil inducción de enzimas metabolizadoras de los fármacos o débil inhibición de la anhidrasa carbónica. A pesar de que la toxicidad materna y paterna fue tan baja como 8 mg/kg/día, no se observaron efectos sobre la fertilidad en ratas machos o hembras con dosis de hasta 100 mg/kg/día. En estudios preclínicos, el topiramato ha mostrado tener efectos teratogénicos en las especies estudiadas (ratones, ratas y conejos). En ratones los pesos fetales y la osificación esquelética fueron reducidos a 500 mg/kg/día junto con la toxicidad materna. Los números globales de malformaciones fetales en ratones fueron aumentadas para todos los grupos tratados con el fármaco (20, 100 y 500 mg/kg/día), pero no se observaron diferencias significativas o relaciones dosis-respuesta para malformaciones generales o específicas, sugiriendo que pueden estar involucrados otros factores como la toxicidad materna. En ratas, la toxicidad materna relacionada con la dosis y la toxicidad embrio/fetal (pesos fetales reducidos y/u osificaciones esqueléticas) fueron observadas con 20 mg/kg/día con efectos teratogénicos (defectos de extremidades y dígitos) a 400 mg/kg/día y superiores. En conejos, la toxicidad materna relacionada con la dosis se advirtió con menos de 10 mg/kg/día con toxicidad embriofetal (letalidad aumentada) con menos de 35 mg/kg/día, y efectos teratogénicos (malformaciones de costillas y vértebras) a 120 mg/kg/día. Los efectos teratogénicos observados en ratas y conejos fueron similares a aquellos observados con los inhibidores de anhidrasa carbónica, que no se han asociado con malformaciones en humanos. Los efectos en el crecimiento también fueron indicados por pesos bajos al nacer, y durante la lactancia para crías de ratas hembra tratadas con 20 ó 100 mg/kg/día durante la gestación y lactancia. En ratas, el topiramato cruza la barrera placentaria. En una batería de ensayos in vitro e in vivo, el topiramato no muestra potencial genotóxico.
Dosis y vía de administración: Oral. Se recomienda que la terapia sea iniciada a dosis baja seguida de una titulación para lograr una dosis efectiva. Topiramato se encuentra disponible en tabletas. Se recomienda no partir las tabletas. No es necesario monitorear las concentraciones plasmáticas de topiramato para optimizar la terapia con topiramato. En raras ocasiones, la adición de topiramato a fenitoína puede requerir de un ajuste de la dosis de fenitoína para alcanzar el óptimo resultado clínico. Puede requerirse ajustar la dosis de topiramato a la adición o retiro de fenitoína y carbamazepina a terapia adjunta con topiramato. Topiramato puede tomarse sin alimentos. Terapia adjunta para epilepsia: Adultos: Se debe iniciar la terapia con 25-50 mg por la noche durante una semana. Se ha reportado el uso de dosis iniciales más bajas, pero no han sido estudiadas sistemáticamente. Subsecuentemente, a intervalos de una o dos semanas, la dosis debe ser incrementada en 25-50 [a 100] mg/día y tomarse en 2 dosis divididas. La titulación de la dosis se debe guiar por la evaluación clínica. Algunos pacientes pueden alcanzar la eficacia con una sola dosis al día. En estudios clínicos de terapia adjunta, 200 mg fueron efectivos siendo la dosis más baja estudiada. Por lo tanto, se considera la dosis mínima efectiva. La dosis usual diaria es 200-400 mg en dos dosis divididas. Pacientes individuales han recibido dosis tan altas como 1,600 mg/día. Ya que topiramato se retira del plasma por hemodiálisis, se deberá administrar una dosis suplementaria de topiramato igual a aproximadamente la mitad de la dosis diaria los días de hemodiálisis. La dosis suplementaria se deberá administrar en dosis divididas al inicio y a la terminación del procedimiento de hemodiálisis. La dosis suplementaria puede diferir basándose en las características del equipo de diálisis utilizado. Estas recomendaciones de dosificación aplican a todos los adultos incluyendo ancianos, en ausencia de enfermedad renal subyacente. Niños mayores de 2 años: La dosis total recomendada de topiramato como terapia adjunta es aproximadamente de 5 a 9 mg/kg/día en 2 dosis divididas. La titulación deberá empezar con 25 mg (o menos, basados en el rango de 1 a 3 mg/kg/día) cada noche durante la primera semana. Después la dosis será incrementada en intervalos de 1 a 2 semanas con aumentos de 1 a 3 mg/kg/día (administrados en 2 dosis divididas), para alcanzar la respuesta clínica óptima. El incremento de la dosificación debe guiarse por los efectos clínicos. Se han estudiado dosis hasta 30 mg/kg/día y fueron generalmente bien toleradas. Monoterapia en epilepsia general: Cuando se retiran otros medicamentos antiepilépticos (MAE) concomitantes para alcanzar la monoterapia con topiramato, se debe tener consideraciones para los efectos que esto puede tener en el control de la crisis. A menos que por cuestiones de seguridad se requiera un retiro abrupto del MAE concomitante, se recomienda retirar gradualmente en una proporción aproximada de una tercera parte de la dosis del MAE concomitante cada 2 semanas. Cuando se retiran fármacos que inducen enzimas, los niveles de topiramato se incrementan. Se pueden requerir disminuir la dosis de topiramato si se indica clínicamente. Adultos: La titulación deberá iniciar con una dosis de 25 mg cada noche por una semana, después la dosis puede ser incrementada en intervalos de 1-2 semanas con aumentos de 25 ó 50 mg/día en 2 dosis divididas. Si el paciente está indispuesto para tolerar el régimen de titulación, se pueden usar incrementos menores o intervalos más largos. El incremento de la dosis y de la titulación se debe guiar por los efectos clínicos. La dosis inicial ideal recomendada para la monoterapia con topiramato en adultos es de 100 mg/día y la dosis diaria máxima recomendada es 500 mg. Algunos pacientes con formas refractarias de epilepsia han tolerado la monoterapia con topiramato a dosis de 1,000 mg/día. Estas dosis recomendadas aplican a todos los adultos incluyendo pacientes de edad avanzada, sin antecedentes de enfermedad renal. Niños: El tratamiento para niños de 2 años en adelante, se debe iniciar con 0.5 a 1 mg/kg por las noches durante la primera semana. Después la dosis se puede incrementar en intervalos de 1 ó 2 semanas con aumentos de 0.5 a 1 mg/kg/día, administrados en 2 dosis divididas. Si el niño está indispuesto para tolerar el régimen de titulación, se pueden usar incrementos menores o intervalos más largos. Se debe guiar la dosis y la titulación de la dosis por evaluación clínica. El rango de dosis recomendada para iniciar la monoterapia con topiramato en niños de 2 años o más es 3 a 6 mg/kg/día. Niños en los que recientemente se diagnosticó crisis de inicio parcial recibieron dosis hasta 500 mg/día. Migraña: La dosis total diaria recomendada de topiramato para la profilaxis de cefalea migrañosa es de 100 mg/día administrada en dos dosis divididas. La titulación deberá iniciar a 25 mg/kg por las noches durante una semana. Después la dosis se puede incrementar en 25 mg/día administrados en intervalos de 1 semana. Si el paciente está indispuesto para tolerar el régimen de titulación, se pueden utilizar intervalos más largos entre los ajustes de dosis, administrados en 2 dosis divididas. Si el niño está indispuesto para tolerar el régimen de titulación, se pueden usar incrementos menores o intervalos más largos. Algunos pacientes pueden experimentar un beneficio con una dosis total diaria de 50 mg/día. Los pacientes han recibi