TRUVADA®
STENDHAL
Denominación genérica: Emtricitabina y tenofovir disoproxil fumarato.
Forma farmacéutica y formulación: Tabletas recubiertas: tenofovir disoproxil fumarato 300 mg (equivalente a 245 mg de tenofovir disoproxil), emtricitabina 200 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: TRUVADA® es una asociación de EMTRIVA® y VIREAD®. Está indicado, en combinación con otros agentes antirretrovirales (tales como los inhibidores no nucleósidos de la transcriptasa reversa o los inhibidores de proteasa), para el tratamiento de la infección por el VIH-1 en adultos. Información adicional importante respecto del empleo de TRUVADA® para el tratamiento de la infección VIH-1: no se recomienda emplear TRUVADA® como componente de un régimen triple de análogos nucleósidos. No se debe coadministrar TRUVADA® con ATRIPLA™ (una asociación de dosis fija de efavirenz, emtricitabina y tenofovir disoproxil fumarato), EMTRIVA®, VIREAD® ni productos que contengan lamivudina (véase Precauciones generales). En pacientes con tratamiento previo, el uso de TRUVADA® debe basarse en los analisis de laboratorio y tratamientos previos (véase Farmacocinetica y Farmacodinamia en humanos). Estudios clínicos: el estudio clínico 934 avala el uso de tabletas de TRUVADA® para el tratamiento de la infección por VIH-1. Los datos adicionales que avalan el uso de TRUVADA® se obtuvieron del estudio 903, el que se administraron lamivudina y tenofovir disoproxil fumarato (tenofovir DF) juntos a pacientes adultos sin tratamiento previo, y del estudio clínico 303, en el que la emtricitabina y la lamivudina demostraron patrones de eficacia, seguridad y resistencia comparables como parte de regímenes multifármaco. Para obtener información adicional sobre estos estudios, consulte la información de prescripción de tenofovir DF y emtricitabina. Estudio 934: se informan los datos obtenidos tras 144 semanas del estudio 934, un estudio multicéntrico, aleatorizado, abierto, control activo, en que se comparó emtricitabina + tenofovir DF, administrados en asociación con efavirenz frente a la asociación en dosis fijas de zidovudina y lamivudina, asociadas a efavirenz, en 511 pacientes sin tratamiento previo con antirretrovirales. De 96a a 144ª semanas del estudio, los pacientes recibieron TRUVADA® con efiverenz en lugar de emtricitabina + tenofovir DF con efavirenz. Los pacientes tenían una media de edad de 38 años (límites, 18 a 80 años), 86% eran varones, el 59% eran de raza blanca y 23% de raza negra. El recuento promedio inicial de linfocitos CD4+ fue de 245 cels/mm3 (límites 2-1191) y la media de ARN del VIH-1 inicial en el plasma fue de 5,01 log10 copias/ml (límites 3,56 a 6,54). Los pacientes de estratificaron según el recuento inicial de linfocitos CD4+ ( < o ≥200 linfocitos/mm3); el 41% tenía recuentos de linfocitos CD4+ < 200 linfocitos/mm3, y el 51% de los pacientes tenía cargas víricas iniciales > 100.000 copias/ml. En la tabla 1 se presentan los resultados del tratamiento después de 48 y de 144 semanas de tratamiento, en los pacientes que no presentaban resistencia al efavirenz al inicio: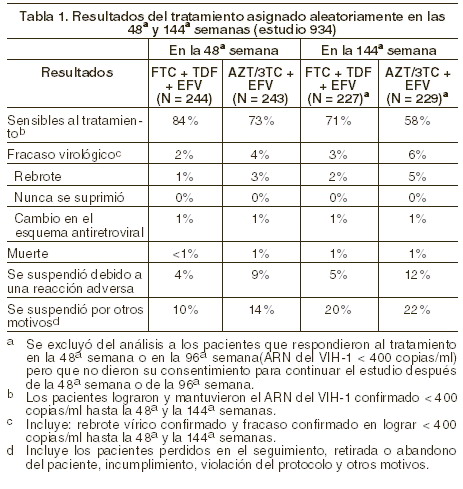
Hasta la 48ª semana, el 84% de los pacientes del grupo tratado con emtricitabina + tenofovir DF y el 73% de los pacientes tratados con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml (hasta la 144ª semana: 71% y 58%, respectivamente). En este estudio abierto, la diferencia en la proporción de pacientes que lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml después de 48 semanas de tratamiento es principalmente el resultado del mayor número de suspensiones debidas a reacciones adversas y a otros motivos en el grupo tratado con zidovudina y lamivudina. Además, el 80% de los pacientes del grupo tratado con emtricitabina + tenofovir DF y el 70% de los pacientes tratados con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 50 copias/ml hasta la 48ª semana (hasta la 144ª semana: 64% y 56%, respectivamente). En la 48ª semana, el aumento medio con respecto a los valores iniciales del recuento de linfocitos CD4+ fue de 190 linfocitos/mm3 en el grupo tratado con emtricitabina + tenofovir DF, y de 158 linfocitos/mm3 en el grupo que recibió zidovudina y lamivudina (en la 144ª semana: 312 y 271 linfocitos/mm3, respectivamente). A las 48 semanas, siete pacientes del grupo tratado con emtricitabina + tenofovir DF y cinco pacientes del grupo tratado con zidovudina y lamivudina experimentaron una nueva reacción de clase C, según el código de los CDC (10 y 6 pacientes, respectivamente, hasta las 144 semanas).
Farmacocinética y farmacodinamia: Las tabletas de TRUVADA® contienen una combinación de emtricitabina y tenofovir disoproxil fumarato en una dosis fija. EMTRIVA® es el nombre comercial de la emtricitabina, un análogo nucleósido sintético de la citidina. El tenofovir disoproxil fumarato (tenofovir DF) es convertido in vivo en tenofovir, un análogo (nucleótido) fosfonato nucleósido acíclico de la 5'-monofosfato adenosina. Tanto la emtricitabina como el tenofovir exhiben actividad inhibidora contra la transcriptasa reversa del VIH-1. Emtricitabina: el nombre químico de la emtricitabina es 5-fluoro-1-(2R,5S)-[2-(hidroximetil)-1,3-oxatiolan-5-il]citosina. La emtricitabina es el enantiómero (-) de un análogo tío de la citidina, el cual se diferencia de otros análogos de la citidina en que tiene una fluorina en la posición 5. La emtricitabina tiene una fórmula molecular de C8H10FN3O3S y un peso molecular de 247,24. Tiene la siguiente fórmula estructural: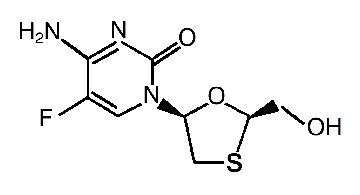
La emtricitabina es un polvo cristalino blanco a incoloro con una solubilidad de 112 mg/ml en agua a 25°C. El coeficiente de partición (Log p) para la emtricitabina es -0,43 y el pKa es de 2,65. Tenofovir disoproxil fumarato: el tenofovir disoproxil fumarato es una sal de ácido fumárico del bis-isopropoxicarbonilmetil éster derivado del tenofovir. El nombre químico del tenofovir disoproxil fumarato es 9-[(R)-2[[bis[[(isopropoxicarbonilo)oxil]-metoxi]fosfinil]metoxil]propil]fumarato de adenina (1:1). Este tiene una fórmula molecular de C19H30N5O10P•C4H4O4 y un peso molecular de 635,52. Tiene la siguiente fórmula estructural: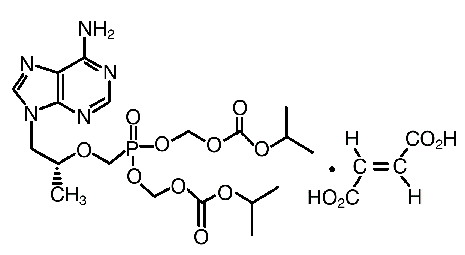
El tenofovir disoproxil fumarato es un polvo cristalino blanco a incoloro con una solubilidad de13,4 mg/ml en agua a 25°C. El coeficiente de participación (Log p) para el disoproxil tenofovir es de 1,25 y el pKa es de 3,75. Todas las dosificaciones se expresan en términos del tenofovir disoproxil fumarato, excepto cuando se hace notar lo contrario. Las tabletas de TRUVADA® se administran por vía oral. Cada tableta recubierta contiene 200 mg de emtricitabina y 300 mg de tenofovir disoproxil fumarato (que equivalen a 245 mg de tenofovir disoproxil) como ingredientes activos. Las tabletas también incluyen los siguientes ingredientes inactivos: croscarmelosa sódica, monohidrato de lactosa, estearato de magnesio, celulosa microcristalina y almidón pregelatinizado (sin gluten). Las tabletas están cubiertas con OPADRY II azul Y-30-10701, el cual contiene laca de aluminio azul #2 FD&C, hidroxipropil metilcelulosa 2910, monohidrato de lactosa, dióxido de titanio y triacetin. Para obtener la información adicional sobre el mecanismo de acción, la actividad antiviral, la resistencia y la resistencia cruzada, consulte la información de prescripción de EMTRIVA® y VIREAD®. Mecanismo de acción: TRUVADA® es una asociación de dosis fijas de fármacos antirretrovirales, emtricitabina y tenofovir disoproxil fumarato. Emtricitabina: la emtricitabina, un análogo nucleósido sintético de la histidina, es fosforilada por enzimas celulares para formar la 5'-trifosfato emtricitabina. La 5'-trifosfato emtricitabina inhibe la actividad de la transcriptasa reversa (TR) del VIH-1 por competencia con el sustrato natural 5'-trifosfato desoxicitidina y es incorporada al ADN viral incipiente, lo que resulta en la terminación de la cadena. La 5'-trifosfato emtricitabina es un inhibidor débil de las polimerasas a, b, e del ADN de los mamíferos y de la polimerasa c del ADN mitocondrial. Tenofovir disoproxil fumarato: el tenofovir disoproxil fumarato es un diéster fosfonato nucleósido acíclico análogo del monofosfato de adenosina. El tenofovir disoproxil fumarato requiere hidrólisis diéster inicial para su conversión a tenofovir y fosforilaciones subsecuentes por las enzimas celulares para formar difosfato de tenofovir. El difosfato de tenofovir inhibe la actividad de la TR del VIH-1 por competencia con el substrato natural 5'-trifosfato desoxiadenosina y, una vez incorporado al ADN, por su terminación. El difosfato de tenofovir es un inhibidor débil de las ADN-polimerasas a, b y de la ADN-polimerasa mitocondrial c en mamíferos. Actividad antiviral: emtricitabina y tenofovir disoproxil fumarato: en estudios de combinación para evaluar la actividad antiviral en cultivo celular de la combinación de emtricitabina y tenofovir, se observaron efectos antivirales sinérgicos. Emtricitabina: se valoró la actividad antiviral de la emtricitabina contra cepas de laboratorio y clínicas del VIH, en líneas de células linfoblastoides, en la línea celular MAGI-CCR5 y en células mononucleares de sangre periférica. Los valores de la concentración eficaz al 50% (CE50) para la emtricitabina estuvieron dentro del rango de 0,0013-0,64 mM (0,0003-0,158 mg/ml). En estudios de combinación de la emtricitabina con otros inhibidores nucleósidos de la transcriptasa reversa (abacavir, lamivudina, estavudina, zalcitabina, zidovudina), con inhibidores no nucleósidos de la transcriptasa reversa (delavirdina, efavirenz, nevirapina), o con inhibidores de proteasa (amprenavir, nelfinavir, ritonavir, saquinavir), se observaron efectos de aditivos a sinérgicos. La emtricitabina exhibió actividad antiviral en cultivo celular contra el VIH-1 tipos A, B, C, D, E, F y G (los valores de la CE50 variaron de 0,007-0,075 mM) y mostró actividad específica contra el VIH-2 (los valores de la CE50 variaron de 0,007-1,5mM). Tenofovir disoproxil fumarato: se valoró la actividad antiviral de tenofovir contra cepas de laboratorio y clínicas del VIH-1 en líneas de células linfoblastoides, en células de monolitos/macrófagos primarios y en linfocitos de sangre periférica. Los valores de la CE50 para tenofovir estuvieron en el intervalo de 0,04-8,5 mM. En los estudios de combinación del tenofovir con los inhibidores nucleósidos de la transcriptasa reversa (abacavir, didanosina, lamivudina, estavudina, zalcitabina, zidovudina), con inhibidores no nucleósidos de la transcriptasa reversa (delavirdina, efavirenz, nevirapina), o con inhibidores de proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir), se observaron efectos de aditivos a sinérgicos. Tenofovir exhibió actividad antiviral en cultivo celular contra VIH-1 tipos A, B, C, D, E, F, G y O (los valores de la CE50 variaron de 0,5-2,2 mM) y actividad específica de cepas contra el VIH-2 (los valores de la CE50 fueron de entre 1,6 mM y 4,9 mM). Resistencia: emtricitabina y tenofovir disoproxil fumarato: se han seleccionado en un cultivo celular cepas de VIH-1 con susceptibilidad reducida a la combinación de emtricitabina y tenofovir. Mediante el análisis del genotipo de estas cepas, se identificaron sustituciones del aminoácido M184I/V y/o del K65R en la transcriptasa reversa viral. En un estudio clínico con pacientes sin tratamiento previo (estudio 934, ver Indicaciones terapéuticas, Estudios clínicos), se realizó un análisis de resistencia en inóculos de VIH-1 de todos los pacientes que habían experimentado fracaso virológico confirmado > 400 copias/ml de ARN del VIH-1 a la semana 144 o que habían abandonado el estudio en forma prematura. El desarrollo de sustituciones asociadas con la resistencia al efavirenz fuer muy frecuente y similar en todos los grupos de tratamiento. Se observó la sustitución de aminoácidos M184V, asociada con resistencia al EMTRIVA® y lamivudina, en 2/19 de los inóculos aislados de pacientes analizados en el grupo tratado con EMTRIVA® + VIREAD®, y en 10/29 de los inóculos asilados de los pacientes analizados en grupo tratado con zidovudina y lamivudina. En las 144 semanas del estudio 934, ninguno de los pacientes que participan en el estudio 934 han desarrollado una sustitución K65R detectable en su VIH-1 según un análisis genotípico estándar. Emtricitabina: se han aislado, en cultivo celular e in vivo, cepas de VIH resistentes a la emtricitabina. El análisis genotípico de estos aislados reveló que la susceptibilidad reducida a la emtricitabina se asocia con una mutación en el codón 184 del gen de la TR del VIH, lo cual resultó en una sustitución del aminoácido metionina por valina o isoleucina (M184V/I). Tenofovir disoproxil fumarato: se han aislado en un cultivo celular cepas del VIH-1 con susceptibilidad reducida al tenofovir. Estos virus expresaron una mutación K65R en la TR y mostraron una reducción de 2-4 veces en la susceptibilidad al tenofovir. En pacientes naïve, los inóculos de 8/47 (17%) pacientes analizados desarrollaron la sustitución K65R en el grupo tratado con VIREAD® después de 144 semanas; 7 casos se produjeron en las primeras 48 semanas de tratamiento y 1 en la semana 96. En pacientes ya previamente tratados con otros fármacos antirretrovirales 14/304 (5%) cepas aisladas de pacientes que tuvieron falla virológica hasta las 96 semanas mostraron una susceptibilidad reducida al tenofovir > 1,4 veces (mediana 2,7). El análisis genotípico de estas cepas resistentes reveló una mutación en el gen de la TR del VIH-1 que resulta en la sustitución del aminoácido K65R. Resistencia cruzada: emtricitabina y tenofovir disoproxil fumarato: se ha detectado resistencia cruzada con ciertos inhibidores nucleósidos de la transcriptasa reversa. Las sustituciones M184V/I y/o K65R seleccionadas en cultivo celular con la combinación de emtricitabina y tenofovir también se observaron en algunos aislados del VIH-1 de sujetos que tuvieron falla al tratamiento con tenofovir en combinación ya sea con lamivudina o con emtricitabina y también con abacavir o didanosina. Por consiguiente, puede ocurrir resistencia cruzada entre estos fármacos en pacientes cuyo virus contenga una o ambas sustituciones de estos aminoácidos. Emtricitabina: los aislados resistentes a la emtricitabina (M184V/I) tuvieron resistencia cruzada a lamivudina y zalcitabina pero conservaron la susceptibilidad in vitro a didanosina, estavudina, tenofovir, zidovudina y a los inhibidores no nucleósidos de la TR (delavirdina, efavirenz y nevirapina). Las cepas del VIH-1 que contienen la sustitución K65R, seleccionada in vivo con abacavir, didanosina, tenofovir y zalcitabina mostraron susceptibilidad reducida a la emtricitabina. Los virus que contienen sustituciones que confieren susceptibilidad reducida a estavudina y zidovudina (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) o didanosina (L74V) permanecieron sensibles a la emtricitabina. El VIH-1 que contiene la sustitución K103N asociada con la resistencia a los inhibidores no nucleósidos de la TR fue sensible a la emtricitabina. Tenofovir disoproxil fumarato: las cepas del VIH-1 de pacientes (N=20) cuyo virus expresó una media de 3 sustituciones de aminoácido en la TR asociadas a zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) mostraron una disminución de 3,1 veces en la susceptibilidad al tenofovir. El VIH-1 resistente a múltiples nucleósidos con una inserción doble T69S en la TR mostró susceptibilidad reducida al tenofovir. Farmacología clínica: farmacocinética: TRUVADA®: la administración de una tableta de TRUVADA® es bioequivalente, en sujetos sanos en ayunas (N=39), a una cápsula de EMTRIVA® (200mg) más una tableta de VIREAD® (300 mg). Emtricitabina: en la tabla 2 se resumen las propiedades farmacocinéticas de emtricitabina. Después de la administración oral de EMTRIVA®, la emtricitabina se absorbe rápidamente y las concentraciones pico en plasma ocurren 1-2 horas postdosis. La unión in vitro de la emtricitabina a las proteínas del plasma humano es menor al 4% y es independiente de la concentración en el rango entre 0,02-200 mg/ml. Después de la administración de emtricitabina radiomarcada, se recuperó aproximadamente el 86% en la orina y el 13% se recuperó como metabolitos. Los metabolitos de emtricitabina incluyen los 3'diastereómeros sulfóxido y su conjugado de ácido glucurónico. La emtricitabina es eliminada mediante una combinación de filtración glomerular y secreción tubular activa. Después de una sola dosis oral de EMTRIVA®, la vida media de emtricitabina en plasma es aproximadamente de 10 horas. Tenofovir disoproxil fumarato: las propiedades farmacocinéticas del tenofovir disoproxil fumarato se resumen en la tabla 2. Después de la administración oral de VIREAD®, las concentraciones máximas de tenofovir en suero se consiguen en 1,0 ± 0,4 horas. La unión in vitro de tenofovir a las proteínas del plasma humano es menor de 0,7% y es independiente de la concentración entre los límites de 0,01-25 mg/ml. Aproximadamente 70-80% de la dosis intravenosa de tenofovir se recuperó como fármaco sin cambios en la orina. Tenofovir se elimina mediante una combinación de filtración glomerular y de secreción tubular activa. Después de una sola dosis oral de VIREAD®, la semivida de eliminación terminal de tenofovir es de 17 horas aproximadamente.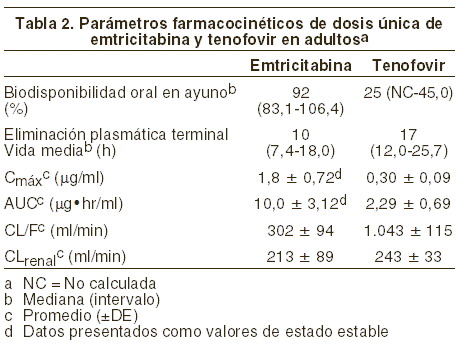
Efectos de los alimentos sobre la absorción oral: TRUVADA® se puede administrar con o sin alimentos. La administración de TRUVADA® después de una comida alta en grasas (784 kcal; 49 gramos de grasas) o de una comida ligera (373 kcal; 8 gramos de grasas) retardó el tiempo de la Cmáx de tenofovir en aproximadamente 0,75 horas. La media del incremento del ABC y de la Cmáx de tenofovir fue de 35% y 15% respectivamente, cuando se administró con una comida alta en grasas o con una comida ligera, comparada con la administración en estado de ayuno. En los estudios previos de seguridad y eficacia, VIREAD® (tenofovir) se tomó en condiciones de haber ingerido alimentos. La exposición sistémica a emtricitabina (ABC y Cmáx) no fue afectada cuando se administró TRUVADA® ya sea con una comida alta en grasas o con una comida ligera. Poblaciones especiales: raza: emtricitabina: no se detectaron diferencias farmacocinéticas debidas a la raza, luego de la administración de EMTRIVA®. Tenofovir disoproxil fumarato: la cantidad de sujetos de grupos raciales y étnicos aparte del grupo de raza blanca reclutados en los estudios clínicos fue insuficiente para determinar adecuadamente las diferencias farmacocinéticas potenciales entre estas poblaciones después de la administración de VIREAD®. Género: las farmacocinéticas de emtricitabina y tenofovir disoproxil fumarato son similares en los pacientes masculinos y femeninos. Pacientes pediátricos y geriátricos: no se han realizado estudios farmacocinéticos de tenofovir en pacientes pediátricos ( < 18 años). No se han evaluado completamente las farmacocinéticas de emtricitabina y tenofovir en ancianos (mayores de 65 años). Pacientes con daño de la función renal: las farmacocinéticas de emtricitabina y tenofovir se alteran en los pacientes con daño renal (véase Precauciones generales). En los pacientes con depuración de creatinina menor de 50 ml/min, la Cmáx y el ABC0-∞ de la emtricitabina y de tenofovir se incrementaron. Se recomienda modificar el intervalo de la dosis de TRUVADA® en los pacientes con depuración de creatinina de 30-49 ml/min. No debe administrarse TRUVADA® a pacientes con depuración de creatinina menor de 30 ml/min ni a pacientes con enfermedad renal en etapa terminal que requieren diálisis (véase Dosis y vía de administración). Pacientes con daño hepático: se han estudiado las farmacocinéticas de tenofovir luego de una dosis de 300 mg de VIREAD® en pacientes no infectados por el VIH con daño hepático moderado a severo. No hubo alteraciones sustanciales en las farmacocinéticas de tenofovir en los pacientes con daño hepático, comparados con los pacientes sin daño hepático. Las farmacocinéticas de TRUVADA® o emtricitabina no han sido estudiadas en pacientes con daño hepático; sin embargo, la emtricitabina no es metabolizada significativamente por las enzimas del hígado, por lo que la repercusión del daño hepático debe ser limitada.
Contraindicaciones: TRUVADA® está contraindicado en los pacientes que previamente han mostrado hipersensibilidad a alguno de los componentes del producto.
Precauciones generales: Acidosis láctica/hepatomegalia severa con esteatosis: se han reportado casos de acidosis láctica y hepatomegalia severa con esteatosis, incluyendo algunos casos de muerte con el empleo de los análogos nucleósidos solos o en combinación con otros antirretrovirales. La mayoría de estos casos se ha observado en mujeres. La obesidad y la exposición prolongada a los nucleósidos pueden ser factores de riesgo. Debe tenerse especial precaución cuando se administran análogos nucleósidos a un paciente con factores de riesgo conocidos para enfermedad del hígado; sin embargo, también se han reportado casos en pacientes sin factores de riesgo conocidos. El tratamiento con TRUVADA® debe suspenderse en cualquier paciente que presenta datos clínicos o de laboratorio sugestivos de acidosis láctica o de hepatotoxicidad importante (entre los cuales se puede incluir hepatomegalia y esteatosis aún en ausencia de elevaciones importantes de transaminasas). Pacientes con infección concomitante por VIH y virus de la hepatitis B (VHB): se recomienda que todos los pacientes con VIH-1 sean examinados en busca del virus de la hepatitis B (VHB) crónica antes de iniciar el tratamiento antirretroviral. TRUVADA® no está aprobado para el tratamiento de la infección crónica por el virus de la hepatitis B y no se ha establecido la seguridad y la eficacia de TRUVADA® en pacientes infectados concomitantemente con VHB y VIH-1. Se han reportado exacerbaciones agudas graves de la hepatitis B en pacientes coinfectados con VHB y VIH-1 que han suspendido la administración de EMTRIVA® o VIREAD®. En algunos de estos pacientes infectados por el VHB y tratados con EMTRIVA®, las exacerbaciones de hepatitis B se asociaron con descompensación hepática e insuficiencia hepática. La función hepática debe monitorearse de manera muy estrecha con observación clínica y pruebas de laboratorio, por lo menos durante varios meses en aquellos pacientes que están infectados concomitantemente con VIH y VHB y que discontinúan TRUVADA®. Si es conveniente, se puede justificar el inicio del tratamiento para la hepatitis B. Nueva aparición o empeoramiento de la disfunción renal: emtricitabina y tenofovir se eliminan principalmente por el riñón. Se ha reportado daño renal, incluso casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia severa), asociados con el uso de VIREAD® (véase Reacciones secundarias y adversas, Experiencia postcomercialización). Se recomienda calcular la eliminación de creatinina en todos los pacientes antes de iniciar la terapia y según se requiera clínicamente durante el tratamiento con TRUVADA®. Debe realizarse el monitoreo rutinario de la eliminación de creatinina calculada y el fósforo sérico en todos los pacientes con riesgo de trastornos renales. Se recomienda el ajuste del intervalo de dosis de TRUVADA® y el estricto monitoreo de la función renal en todos los pacientes con eliminación de creatinina de 30 a 49 ml/min (véase Dosis y vía de administración). No hay datos de seguridad ni eficacia disponibles sobre pacientes con disfunción renal que hayan recibido TRUVADA® siguiendo estas pautas de dosificación, por lo que el beneficio potencial de la terapia con TRUVADA® debe evaluarse en relación con el riesgo potencial de toxicidad renal. No se debe administrar TRUVADA® a pacientes con eliminación de creatinina < 30 ml/min o pacientes que necesiten hemodiálisis. Debe evitarse tomar TRUVADA® concurrentemente con medicamentos nefrotóxicos o luego de haber tomado estos medicamentos en fecha reciente. Administración concomitante de productos relacionados: TRUVADA® es una combinación de dosis fijas de emtricitabina y tenofovir disoproxil fumarato. TRUVADA® no debe administrarse en forma concomitante con ATRIPLATM, EMTRIVA® o VIREAD®. Debido a la similitud entre emtricitabina y lamivudina, TRUVADA® no debe administrarse en forma concomitante con otros medicamentos que contengan lamivudina entre los que se incluye Combivir™ (lamivudina/zidovudina), Epivir™ o Epivir™ -VHB (lamivudina), Epzicom™ (abacavir sulfato/lamivudina) o Trizivir™ (abacavir sulfato/lamivudina/zidovuldina). Disminución de la densidad ósea: se debe planear la vigilancia de la densidad ósea para los pacientes infectados por el VIH-1 que tienen antecedentes de fracturas óseas patológicas o con riesgo de osteopenia. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos para todos los pacientes. Se debe obtener asesoramiento adecuado si se sospecha de la presencia de anomalías óseas. Tenofovir disoproxil fumarato: en un estudio de 144 semanas de duración, en pacientes sin tratamiento previo con antirretrovirales, se observaron disminuciones en la densidad mineral ósea (DMO) en columna lumbar y en la cadera, en ambos grupos de tratamiento del estudio. En la semana 144, hubo una disminución en el porcentaje medio significativamente superior respecto al valor inicial de DMO de la columna lumbar en los pacientes tratados con VIREAD® + lamivudina + efavirenz en comparación con los pacientes tratados con estavudina + lamivudina + efavirenz. Los cambios en la DMO en la cadera fueron similares en los dos grupos de tratamiento. En ambos grupos, la mayor parte de la disminución de la DMO se produjo en las primeras 24 a 48 semanas del estudio y se mantuvo durante 144 semanas. El 28% de los pacientes tratados con VIREAD® frente al 21% de los pacientes del grupo de comparación experimentaron una disminución de al menos 5% de la DMO en la columna o del 7% de la DMO en la cadera. Se notificaron fracturas clínicamente relevantes (sin incluir los dedos de los pies y las manos) en cuatro pacientes del grupo de tratado con VIREAD® y en seis pacientes del grupo de comparación. El tenofovir disoproxil fumarato se asoció con aumentos significativos en los marcadores bioquímicos del metabolismo óseo (fosfatasa alcalina sérica específica del tejido óseo, osteocalcina sérica, telopéptido C sérico y telopéptido N urinario), lo que sugiere un mayor recambio óseo. Las concentraciones de hormona paratiroidea en el suero y de 1,25-vitamina D también fueron más altas en los pacientes tratados con VIREAD®. Se desconocen los efectos de los cambios asociados a VIREAD® en la DMO y de los marcadores bioquímicos en la salud ósea a largo plazo y en el riesgo futuro de fracturas. Para obtener información adicional, consulte la información para prescribir de VIREAD®. Se han notificado casos de osteomalacia (asociada a tubulopatía renal proximal) en relación con el uso de VIREAD® (véase Reacciones secundarias y adversas). Redistribución de la grasa: en pacientes que han recibido tratamiento antirretroviral, se ha observado redistribución/acumulación de grasa del cuerpo, incluida la obesidad central, aumento de la grasa dorso cervical (giba de búfalo), adelgazamiento periférico, facial, crecimiento de las mamas y "aspecto cushinoide". El mecanismo y las consecuencias a largo plazo de estas reacciones se desconocen por ahora. No se ha establecido una relación causal. Síndrome de reconstitución inmunológica: se han informado casos de síndrome de reconstitución inmunológica en pacientes tratados con terapia antirretroviral de combinación, incluidos EMTRIVA® y VIREAD®. Durante la fase inicial del tratamiento antirretroviral de combinación, los pacientes cuyo sistema inmunológico responde pueden desarrollar una respuesta inflamatoria a infecciones oportunistas residuales o indolentes (tales como infección por Mycobaterium avium, citomegalovirus, Pneumocystis jirovecii (PCP) o el bacilo de la tuberculosis, que pueden requerir evaluación y tratamiento adicionales. Toxicología o farmacología en animales: se administró tenofovir y tenofovir disoproxil fumarato a ratas, perros y monos en estudios de toxicología, en exposiciones (basados en las ABCs) mayores o iguales a 6 veces a las observadas en humanos que causaron toxicidad ósea. En los monos, la toxicidad ósea se diagnosticó como osteomalacia. La osteomalacia observada en los monos pareció ser reversible al reducir la dosis o discontinuar el tenofovir. En ratas y perros la toxicidad ósea se manifestó como disminución de la densidad ósea. No se conocen los mecanismos intrínsecos de la toxicidad ósea. Se observaron signos de toxicidad renal en cuatro especies animales. Hubo incrementos en los valores de creatinina sérica, nitrógeno de urea en sangre, glucosuria, proteinuria, fosfaturia y/o calciuria y disminución en el fosfato en suero de diversos grados en esos animales. Estos signos de toxicidad se observaron en exposiciones (basados en el ABCs) de dos a veinte veces mayores que las observadas en humanos. No se conoce la relación de las alteraciones renales, en particular la fosfaturia con la toxicidad ósea. Uso pediátrico: no se recomienda la administración de TRUVADA® a pacientes menores de 18 años, ya que es una tableta de combinación de dosis fijas que contiene un componente de VIREAD® cuya seguridad aún no se ha establecido para pacientes de estas edades. Uso geriátrico: los estudios clínicos con EMTRIVA® o VIREAD® no incluyeron una cantidad suficiente de sujetos mayores de 65 años para determinar si ellos tenían una respuesta diferente a las de los sujetos más jóvenes. Como regla, la dosis para los ancianos debe seleccionarse con cautela, teniendo en mente la mayor frecuencia de disminución de la función hepática, renal o cardíaca en estos pacientes así como las enfermedades concomitantes y otros tratamientos con medicamentos. Información para los pacientes: se debe informar a los pacientes lo siguiente: TRUVADA® no cura la infección por el VIH-1 y los pacientes pueden seguir presentando enfermedades asociadas con esta infección, incluso las infecciones oportunistas. Los pacientes deben permanecer bajo el cuidado de un médico cuando utilizan TRUVADA®; no se ha comprobado que el uso de TRUVADA® reduzca el riesgo de transmisión del VIH-1 a otras personas por medio del contacto sexual o de la contaminación de sangre; se desconocen los efectos a largo plazo de TRUVADA®; las tabletas de TRUVADA® son sólo para administración por vía oral; es importante tomar TRUVADA® en un tratamiento de asociación, con un intervalo de dosificación regular, para evitar que el paciente olvide tomar la dosis; se ha notificado la aparición de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales. El tratamiento con TRUVADA® debe interrumpirse en cualquier paciente que presente síntomas clínicos que indiquen la presencia de acidosis láctica o de hepatotoxicidad pronunciada (incluso náuseas, vómitos, molestias gástricas poco habituales o inesperadas y debilidad); se debe realizar a los pacientes con infección por el VIH-1 la prueba para detectar la presencia del virus de la hepatitis B (VIH) antes de iniciar el tratamiento antirretroviral; se han notificado casos de exacerbación aguda grave de la hepatitis B en los pacientes con infección concomitante por el VHB y el VIH-1 que han interrumpido la administración de EMTRIVA® o VIREAD®; se han notificado casos de disfunción renal, entre ellos, casos de insuficiencia renal aguda y del síndrome de Fanconi en relación con el uso de VIREAD®. Se debe evitar el uso de TRUVADA® con el uso reciente o concomitante de un medicamento nefrotóxico. Puede ser necesario ajustar el intervalo de dosificación de TRUVADA® en los pacientes con disfunción renal (véase Dosis y vía de administración); TRUVADA® no debe administrarse concomitantemente con ATRIPLATM, EMTRIVA® o VIREAD®, ni con fármacos que contengan lamivudina, como Combivir® (lamivudina y zidovudina), Epivir® o Epivir-HBV® (lamivudina), Epzicon® (sulfato de abacavir y lamivudina) o Trizivir® (sulfato de abacavir, lamivudina y zidovudina); TRUVADA® no debe administrarse con HEPSERA (véase Precauciones generales); se ha observado disminución de la densidad ósea con el uso de VIREAD®. Se debe plantear la vigilancia de la densidad ósea en los pacientes que tengan antecedentes de fracturas óseas patológicas o con riesgo de osteopenia.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: categoría B. Emtricitabina: la incidencia de variaciones fetales y malformaciones no se incrementó en los estudios de toxicidad embrionaria fetal efectuados con emtricitabina en ratones, con exposiciones (ABC) aproximadamente 60 veces más altas, y en conejos con exposiciones aproximadamente 120 veces más altas que las observadas en humanos con la dosis diaria recomendada. Tenofovir disoproxil fumarato: los estudios de la reproducción se efectuaron en ratas y en conejos con dosis hasta 14 y 19 veces mayores a las humanas, según las comparaciones de la superficie corporal, y no revelaron evidencia de deterioro de la fertilidad ni daño sobre el feto a causa del tenofovir. Sin embargo, no hay estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre predicen la respuesta en humanos, durante el embarazo TRUVADA® solamente debe emplearse cuando sea estrictamente necesario. Registro de embarazos con antirretrovirales: se invita a los profesionales médicos a que registren a las pacientes que se queden embarazadas en el siguiente sitio de internet: www.kendle.com/registries. Madres que amamantan: los centros para el control y la prevención de las enfermedades recomiendan que las madres infectadas con el VIH no deben amamantar a sus hijos para evitar el riesgo de transmisión postnatal del VIH. Los estudios en ratas han demostrado que tenofovir se secreta en la leche. No se sabe si tenofovir se excreta en la leche humana. Tampoco se sabe si la emtricitabina se excreta en la leche humana. Debido al potencial de transmisión del VIH y al potencial de reacciones adversas serias en los infantes alimentados al seno, debe indicarse a las madres que no amamanten a sus bebés si están recibiendo TRUVADA®.
Reacciones secundarias y adversas: En otros apartados de la información para prescribir, se tratan las siguientes reacciones adversas: acidosis láctica o hepatomegalia grave con esteatosis (véase Precauciones generales). Exacerbaciones agudas y graves de la hepatitis B (véase Precauciones generales). Nueva aparición o empeoramiento de la disfunción renal (véase Precauciones generales). Disminución de la densidad ósea (véase Precauciones generales). Síndrome de reconstitución inmunológica (véase Precauciones generales). Reacciones adversas, experiencia en ensayos clínicos: debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Las reacciones adversas más frecuentes (incidencia ≥10%, cualquier gravedad) que se produjeron en el estudio 934, un estudio clínico con control activo de efavirenz, emtricitabina y tenofovir disoproxil fumarato, consisten en diarrea, náuseas, fatiga, cefalea, mareos, depresión, insomnio, anomalías de los sueños y erupción cutánea. Véase también el cuadro 2 la frecuencia de las reacciones adversas aparecidas con el tratamiento (de 2° a 4° grados) que se produjeron en ≥5% de los pacientes tratados con efavirenz, emtricitabina y tenofovir disoproxil fumarato en este estudio. La decoloración de la piel, manifestada por hiperpigmentación de las palmas de las manos o de las plantas de los pies, generalmente fue leve y asintomática. Se desconoce su mecanismo y su importancia clínica. Estudio 934 - reacciones adversas aparecidas con el tratamiento: en el estudio 934, se administró a 511 pacientes sin tratamiento antirretroviral previo VIREAD® + EMTRIVA® asociado a efavirenz (N=257) o bien zidovudina y lamivudina asociados a efavirenz (N=254). Por lo general, las reacciones adversas observados en este estudio fueron coherentes con las observadas en otros estudios en pacientes con o sin tratamiento previo, tratados con VIREAD® o EMTRIVA® (tabla 3).
Además de las reacciones que se describen más arriba en el estudio 934, otras reacciones adversas que se presentaron en al menos el 5% de los pacientes tratados con EMTRIVA® o VIREAD® junto con otros antirretrovirales en estudios clínicos fueron ansiedad, artralgia, aumento de la tos, dispepsia, fiebre, mialgia, dolor, dolor abdominal, dolor lumbar, parestesia, neuropatía periférica (incluso neuritis periférica y neuropatía), neumonía y rinitis. Reacciones adversas, experiencia postcomercialización: se han detectado las siguientes reacciones adversas durante el empleo posterior a la aprobación de VIREAD®. Debido a que se reportaron voluntariamente por una población de tamaño no conocido, no siempre es posible calcular de manera fiable su frecuencia ni establecer una re