VALPROSID
ARMSTRONG
Denominación genérica: Valproato semisódico.
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Valproato semisódico equivalente a 250 mg y 500 mg de ácido valproico. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: Anticonvulsivante. Epilepsia: VALPROSID® cubren todo el espectro terapéutico de las crisis epilépticas en adultos y niños de 10 años de edad y mayores. En los niños menores de 10 años de edad se debe tener precaución por riesgo elevado de hepatotoxicidad y encefalopatía hiperamonémica en caso de tener alteraciones en el ciclo de la urea. Valprosid® está indicado como monoterapia y terapia complementaria en el tratamiento de pacientes con crisis parciales complejas que ocurren en forma aislada o asociadas con otro tipo de crisis. Valprosid® también está indicado en monoterapia o terapia complementaria para el tratamiento de las crisis de ausencia simple y compleja y de manera complementaria en pacientes con múltiples tipos de crisis que incluyen crisis de ausencias. Se define ausencia simple como una muy breve pérdida del sensorio o de conciencia acompañada por ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. Ausencia compleja es el término usado cuando otros signos también están presentes. Véase Precauciones Generales para la declaración respecto a la disfunción hepática fatal. Migraña: Valprosid® tabletas está indicado para la profilaxis de la cefalea tipo migraña con aura o sin aura, para reducir el número de episodios o la severidad de la crisis. Trastornos del Estado de Ánimo: Constituyen un grupo heterogéneo de enfermedades, típicamente recurrentes, donde se incluyen el trastorno unipolar (depresivo) y el bipolar (maniaco-depresivo). Consisten en alteraciones del humor con carácter infiltrativo, que se acompañan de disfunción psicomotriz y síntomas vegetativos. Manía: Valprosid® está indicado en el tratamiento del trastorno bipolar, en el episodio maniaco agudo y en episodios mixtos asociados al trastorno bipolar, con o sin características psicóticas. En el Trastorno del Estado de Ánimo se incluye el espectro del trastorno bipolar, dentro del cual se encuentra el episodio maniaco que es un estado anormal del ánimo el cual se caracteriza por un estado de ánimo elevado en forma persistente, expansiva e irritable. Así mismo, el episodio mixto está caracterizado por reunir los criterios diagnósticos de un episodio maniaco y un episodio depresivo mayor.
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Absorción/Biodisponibilidad - Dosis equivalentes de valproato semisódico y ácido valproico aportan cantidades equivalentes de ión valproato a nivel sistémico. Aunque la velocidad de absorción del ión valproato puede variar con la formulación administrada (líquido, sólido o sprinkle), con las condiciones de uso (v.gr. ayuno o postprandio), y el método de administración, estas diferencias deben ser de menor importancia clínica bajo condiciones en estado estable alcanzadas con el uso crónico en el tratamiento de epilepsia. Sin embargo, es posible que las diferencias entre los productos de valproato en Tmáx y Cmáx pudieran ser importantes al inicio del tratamiento. Se realizó el Estudio de Bioequivalencia evaluando tanto condiciones de ayuno y posterior a la ingesta de alimentos entre VALPROSID® tabletas con capa entérica conteniendo Valproato Semisódico equivalente a 250 mg de ácido valpróico (Medicamento de Prueba) desarrolladas por Armstrong Laboratorios de México, S.A. de C.V., y EPIVAL® comprimidos con capa entérica conteniendo Valproato Semisódico equivalentes a 250 mg de ácido valpróico (Medicamento de Referencia) fabricados por Abbott Laboratories de México, S.A. de C.V. donde se reportaron los siguientes resultados de sus comportamientos farmacocinéticos: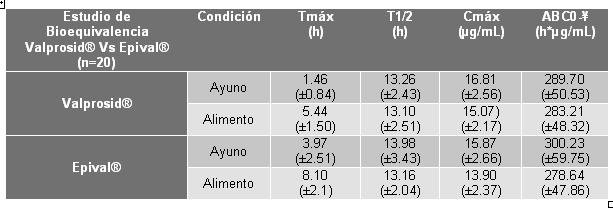
Concluyendo que ambos productos son bioequivalente en condiciones de ayuno y posterior a la ingesta de alimentos. En el estudio de Bioequivalencia entre VALPROSID® tabletas con capa entérica conteniendo 500 mg de Valproato semisódico (Medicamento de prueba) elaboradas por Armstrong Laboratorios de México, S.A. de C.V. y EPIVAL® comprimidos con capa entérica conteniendo 500 mg de Valproato Semisódico (Medicamento de Referencia) fabricado por Abbott Laboratories de México, S.A. de C.V, después de una administración oral de 500 mg se reportan los siguientes resultados en cuanto a su farmacocinética: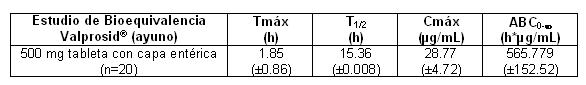
Con los resultados obtenidos en el estudio, se concluye que ambos productos son Bioequivalentes. Mientras la velocidad de absorción desde el aparato G.I. y la fluctuación en las concentraciones plasmáticas de valproato varían con el régimen de dosificación y la formulación, la eficacia de valproato como un anticonvulsivo en el uso crónico es improbable que sea afectado. Con base en la experiencia, empleando dosificaciones de una vez al día a cuatro veces al día, así como en estudios en modelos de epilepsia en primates que involucró una velocidad de infusión constante, indica que la biodisponibilidad sistémica total (grado de absorción) es el principal determinante en el control de las crisis y que las diferencias en los cocientes de las concentraciones plasmáticas pico a valle entre las formulaciones de valproato son intrascendentes desde un punto de vista clínico práctico. La coadministración de productos orales de valproato con alimento y la substitución entre varias formulaciones de valproato semisódico y ácido valproico no debe causar problemas clínicos en el manejo de pacientes con epilepsia (ver Dosis). No obstante, cualquier cambio en dosificación, o la adición o discontinuación de medicamentos concomitantes, debe ser cuidadosamente vigilado el estado clínico y las concentraciones plasmáticas de valproato. Distribución Unión a proteínas: La unión de valproato a proteínas plasmáticas depende de la concentración y la fracción libre aumenta aproximadamente de 10% con 40 mg/mL a 18.5% con 130 mg/mL. La unión a proteínas de valproato disminuye en los ancianos, en pacientes con enfermedades hepáticas crónicas, en pacientes con insuficiencia renal y en presencia de otros fármacos (por ejemplo, ácido acetilsalicílico). A la inversa, valproato puede desplazar ciertos fármacos unidos a proteínas (por ejemplo, fenitoína, carbamazepina, warfarina y tolbutamida) (ver Interacciones medicamentosas). Distribución en el SNC: Las concentraciones de valproato en el líquido cefalorraquídeo (LCR) se aproximan a las concentraciones libres en plasma (alrededor del 10% de la concentración total). Metabolismo: El valproato es metabolizado casi totalmente por el hígado. En pacientes adultos en monoterapia, del 30 al 50% de una dosis administrada aparece en orina como un conjugado glucurónido. La b-oxidación mitocondrial es otra vía metabólica principal, representando normalmente más del 40% de la dosis. Por lo regular, menos de 15 a 20% de la dosis se elimina mediante otros mecanismos oxidativos. Menos del 3% de una dosis administrada se excreta intacta en orina. La relación entre la dosis y la concentración total de valproato es no lineal; la concentración no aumenta proporcionalmente con la dosis, más bien aumenta en menor grado debido a la unión saturable a proteínas plasmáticas. La cinética del fármaco libre es lineal. Eliminación: El promedio de depuración plasmática y el volumen de distribución de valproato total son 0.56 L/h/1.73 m2 y 11 L/1.73 m2, respectivamente. El promedio de depuración plasmática y el volumen de distribución de valproato libre son 4.6 L/h/1.73 m2 y 92 L/1.73 m2, respectivamente. La vida media terminal promedio de la monoterapia con valproato varió de 9 a 16 horas después de los esquemas de dosificación oral de 250 a 1000 mg. Los estimados citados corresponden principalmente a pacientes que no están tomando fármacos que afectan los sistemas de enzimas hepáticas metabólicas. Por ejemplo, los pacientes que toman fármacos antiepilépticos que inducen enzimas (carbamazepina, fenitoína y fenobarbital) eliminarán el valproato más rápidamente. Debido a estos cambios en la depuración de valproato, se debe intensificar el monitoreo de las concentraciones de antiepilépticos siempre que se introduzcan o retiren antiepilépticos concomitantes. Poblaciones especiales: Neonatos: Los niños en los primeros dos meses de vida tienen una capacidad marcadamente reducida para eliminar el valproato en comparación con niños de mayor edad y adultos. Esto es el resultado de una menor depuración (tal vez debido al retraso en el desarrollo de glucuronosiltransferasa y otros sistemas enzimáticos implicados en la eliminación de valproato) así como un mayor volumen de distribución (debido en parte a la menor unión a proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en niños menores de 10 días varió de 10 a 67 horas en comparación con un rango de 7 a 13 horas en niños mayores de dos meses. Niños: Los pacientes pediátricos (es decir, entre 3 meses y 10 años) tienen depuraciones 50% más altas expresadas en peso (es decir, mL/min/kg) que los adultos. Los niños mayores de 10 años tienen parámetros farmacocinéticos que se aproximan a los de los adultos. Ancianos: Se ha demostrado que la capacidad de los pacientes ancianos (rango de edad: 68 a 89 años) para eliminar el valproato es reducida en comparación con la observada en adultos jóvenes (rango de edad: 22 a 26). La depuración intrínseca disminuye 39%; la fracción libre de valproato aumenta en 44%. Por consiguiente, en los ancianos debe reducir la dosis inicial (ver Dosis y vía de administración). Sexo: No hay diferencias en el área de superficie corporal entre los varones y mujeres (4.8±0.17 y 4.7±0.07 L/h por 1.73 m2, respectivamente). Origen étnico: No se han estudiado los efectos del origen étnico en la cinética de valproato. Enfermedad hepática: Ver Contraindicaciones y precauciones generales. La enfermedad hepática deteriora la capacidad de eliminar el valproato. En un estudio, la depuración de valproato libre disminuyó en 50% en siete pacientes con cirrosis y en 16% en cuatro pacientes con hepatitis aguda, en comparación con seis personas sanas. En ese estudio, la vida media de valproato aumentó de 12 a 18 horas. La enfermedad hepática también se relaciona con concentraciones más bajas de albúmina y fracciones libres más grandes de valproato (aumentó de 2 a 2.6 veces). En consecuencia, el monitoreo de las concentraciones totales puede ser engañoso ya que las concentraciones libres pueden ser sustancialmente elevadas en pacientes con enfermedad hepática mientras que las concentraciones totales pueden parecer normales. Enfermedad renal: Se reportó una ligera reducción (27%) en la depuración de valproato libre en pacientes con insuficiencia renal (depuración de creatinina < 10 mL/minuto). Sin embargo, la hemodiálisis normalmente reduce las concentraciones de valproato en alrededor del 20%. Por consiguiente, no parece necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes la unión a proteínas disminuye sustancialmente; por consiguiente, el monitoreo de concentraciones totales puede ser engañoso. Niveles plasmáticos y efecto clínico: La relación entre la concentración plasmática y respuesta clínica no está bien documentada. Un factor que contribuye es la concentración no lineal dependiente de la unión a proteínas de valproato que afecta la depuración del fármaco. Por consiguiente, el monitoreo de valproato sérico total no puede proporcionar un índice confiable de las especies bioactivas de valproato. Por ejemplo, debido a que la unión de valproato a proteínas plasmáticas depende de la concentración, la fracción libre aumenta de aproximadamente 10% con 40 mg/mL a 18.5% con 130 mg/mL. En pacientes geriátricos, con hiperlipidemias o con enfermedades hepáticas y renales se presentan fracciones libres más altas que lo esperado. Epilepsia: El rango terapéutico en epilepsia comúnmente se considera que es 50 a 100 mg /mL de valproato total, aunque algunos pacientes pueden ser controlados con concentraciones plasmáticas más bajas o más altas. Epilepsia: El rango terapéutico en epilepsia comúnmente se considera que es de 50 a 100 mg/mL de valproato total, aunque algunos pacientes pueden ser controlados con concentraciones plasmáticas más bajas o más altas. Propiedades Farmacodinámicas: El valproato semisódico se disocia a ión valproato en el tracto gastrointestinal. El mecanismo de acción por el cual el valproato ejerce su efecto terapéutico no ha sido del todo aclarado. Se sugiere que su actividad en epilepsia está relacionada con un aumento de los niveles cerebrales de ácido gamaminobutírico (GABA).
Contraindicaciones: El valproato semisódico no debe administrarse a pacientes con enfermedad hepática o disfunción hepática significativa. El valproato semisódico está contraindicado en pacientes con hipersensibilidad conocida al medicamento. El valproato semisódico está contraindicado en pacientes con trastorno en el ciclo de la urea (ver Precauciones Generales).
Precauciones generales: Hepatotoxicidad: Se han presentado alteraciones de la función hepática, incluyendo insuficiencia con resultados fatales en algunos pacientes que recibieron ácido valproico. Estos incidentes generalmente han ocurrido durante los primeros seis meses de tratamiento. La hepatotoxicidad severa o fatal puede verse precedida de síntomas inespecíficos como malestar general, debilidad, letargia, edema facial, anorexia y vómito. En pacientes con epilepsia puede ocurrir una pérdida del control de las crisis. Se debe monitorear de cerca a los pacientes para detectar la presencia de estos síntomas. Se deben realizar pruebas de función hepática antes del tratamiento y después en intervalos frecuentes, especialmente durante los primeros seis meses. Sin embargo, los médicos no deben confiar totalmente en la química sanguínea ya que estas pruebas no son anormales en todos los casos, pero también deben considerar los resultados de la historia clínica cuidadosa y exploración física. Se debe tener precaución al administrar productos con valproato semisódico a pacientes con antecedentes de enfermedad hepática. Los pacientes que toman múltiples anticonvulsivos, los niños, los pacientes con trastornos metabólicos congénitos, aquellos con trastornos de crisis convulsivas severas acompañados por retraso mental, y aquellos con enfermedad cerebral orgánica están en riesgo particular. La experiencia indica que los niños menores de dos años tienen un riesgo considerablemente más alto de desarrollar hepatotoxicidad letal, especialmente aquellos con los padecimientos antes mencionados. En este grupo de pacientes, el valproato sódico debe usarse con extrema precaución y como medicamento único. Los beneficios del tratamiento se deben valorar contra los riesgos. Por arriba de este grupo de edad, la experiencia en epilepsia indica que la incidencia de hepatotoxicidad letal disminuye considerablemente en los grupos de pacientes progresivamente mayores. El medicamento se debe suspender de inmediato ante la presencia de disfunción hepática significativa. En algunos casos, la disfunción hepática ha progresado a pesar de suspender el producto. Pancreatitis: Se han reportado casos de pancreatitis potencialmente mortal en niños y adultos tratados con valproato. Algunos de los casos se describieron como hemorrágicos con progresión rápida desde los síntomas iniciales a la muerte. Algunos casos se han presentado brevemente después del uso inicial así como después de varios años de uso. La tasa basada en los casos reportados excede la esperada en la población general y ha habido casos en los que la pancreatitis reincidió después de reanudar el tratamiento con valproato. En estudios clínicos, hubo dos casos de pancreatitis sin etiología alternativa en 2,416 pacientes, representando una experiencia de 1,044 años-paciente. Se debe advertir a los pacientes y cuidadores que el dolor abdominal, náusea, vómito y/o anorexia podrían ser síntomas de pancreatitis que requieren evaluación médica inmediata. Si se diagnostica pancreatitis, el valproato normalmente debe suspenderse. Se debe iniciar un tratamiento alternativo para el padecimiento causante según esté indicado clínicamente. Trastornos del ciclo de la urea (TCU): Se ha reportado encefalopatía hiperamonémica, en ocasiones mortal, después del inicio del tratamiento con valproato en pacientes con trastornos del ciclo de la urea, un grupo de alteraciones genéticas poco comunes, en particular deficiencia de ornitina transcarbamilasa. En los siguientes pacientes debe considerarse la evaluación del TCU antes de iniciar el tratamiento con valproato: 1) pacientes con antecedentes de encefalopatía inexplicada o coma, encefalopatía asociada con el contenido proteico, encefalopatía relacionada con el embarazo o posparto, retraso mental inexplicado o antecedentes de elevaciones plasmáticas de amoniaco o glutamina; 2) pacientes con vómito cíclico o letargo, irritabilidad episódica extrema, ataxia, BUN (Nitrógeno Ureico Sanguíneo) bajo, intolerancia a proteínas; 3) pacientes con antecedentes heredofamiliares de TCU o de muerte del lactante inexplicada (particularmente varones); 4) pacientes con signos o síntomas de TCU. Los pacientes que desarrollan síntomas de encefalopatía hiperamonémica inexplicada mientras reciben tratamiento con valproato deben recibir tratamiento inmediato (incluyendo la suspensión de valproato) y ser evaluados con respecto a trastornos del ciclo de la urea (ver Contraindicaciones y precauciones generales). Comportamiento e ideas de suicidio: Se reportó un mayor riesgo de pensamientos y comportamiento suicidas en pacientes que tomaban fármacos antiepilépticos (FAE) para cualquier indicación. El aumento del riesgo de pensamientos o comportamiento suicidas con fármacos antiepilépticos se observó desde tan solo una semana después de haber iniciado el tratamiento con FAE y persistió durante todo el tratamiento evaluado. El riesgo relativo de pensamientos o comportamiento de suicidio fue más alto en los estudios clínicos de epilepsia que en los estudios clínicos de padecimientos psiquiátricos o de otro tipo, pero las diferencias de riesgo absoluto fueron similares para las indicaciones de epilepsia y psiquiátricas. Se debe monitorear a los pacientes tratados con un FAE para cualquier indicación para detectar la aparición o empeoramiento de depresión, comportamiento o ideas de suicidio y/o cambios inusuales del estado de ánimo o comportamiento. Cuando se prescribe valproato semisódico u algún otro FAE debe valorar el riesgo de comportamiento o pensamientos de suicidio y el riesgo de no tratar la enfermedad. La epilepsia y muchas otras enfermedades para las que se prescriben FAE están asociadas con morbilidad y un mayor riesgo de comportamiento y pensamientos de suicidio. En caso de presentarse pensamientos y comportamientos de suicidio durante el tratamiento, el médico que prescribe debe considerar si la aparición de estos síntomas en cualquier paciente puede estar relacionada con la enfermedad que está tratando. Se debe informar a los pacientes, sus cuidadores y familias que los FAE aumentan el riesgo de pensamientos y comportamiento de suicidio y se les debe advertir de la necesidad de estar alertas de la aparición o empeoramiento de los signos y síntomas de depresión, cualquier cambio inusual del estado de ánimo o comportamiento, o la aparición de pensamientos y comportamientos de suicidio o pensamientos de daño autoprovocado. Los comportamientos de preocupación deben reportarse inmediatamente a su médico. Interacciones con antibióticos carbapenémicos: Los antibióticos carbapenémicos (ertapenem, imipenem, meropenem) pueden reducir las concentraciones séricas de ácido valproico a niveles subterapéuticos, produciendo pérdida del control de crisis convulsivas. Las concentraciones séricas de ácido valproico deben monitorearse frecuentemente después de iniciar el tratamiento con carbapenem. Si las concentraciones séricas de ácido valproico disminuyen significativamente o si el control de crisis convulsivas se deteriora, debe considerarse un tratamiento antibacteriano o anticonvulsivo alternativo (ver Interacciones medicamentosas). Somnolencia en los ancianos: En un estudio doble ciego, multicéntrico de valproato en pacientes de edad avanzada con demencia (la edad promedio era 83 años), las dosis se aumentaron 125 mg/día hasta una dosis de 20 mg/kg/día. Una proporción significativamente más alta de pacientes tratados con valproato tuvieron somnolencia en comparación con los tratados con placebo, y aunque no fue estadísticamente significativa, hubo una proporción más alta de pacientes con deshidratación. También hubo una tasa de suspensión por somnolencia significativamente más alta que con placebo. Algunos pacientes con somnolencia (aproximadamente la mitad), presentaron disminución en la ingesta de alimentos y pérdida de peso. En los pacientes que experimentaron estos eventos hubo una tendencia a tener una concentración basal de albúmina más baja, depuración de valproato más baja y BUN más alto. En pacientes de edad avanzada, la dosis debe aumentarse más lentamente y con un monitoreo periódico de la ingesta de líquidos y alimentos, deshidratación, somnolencia y otros eventos adversos. En pacientes con menor ingesta de alimentos o líquidos y en pacientes con somnolencia excesiva debe considerarse reducir o suspender la dosis de valproato (ver Dosis y vía de administración). Trombocitopenia: La frecuencia de efectos adversos (en particular elevación de enzimas hepáticas y trombocitopenia [véase Precauciones Generales]) puede estar relacionada con la dosis. En un estudio clínico de valproato semisódico como monoterapia en pacientes con epilepsia, 34/126 pacientes (27%) que recibían aproximadamente 50 mg/kg/día en promedio, tuvieron por lo menos un valor de plaquetas ≤ 75 x 109/L. A aproximadamente la mitad de estos pacientes se les suspendió el tratamiento y el número de plaquetas regresó a un nivel normal. En los pacientes restantes, el número de plaquetas se normalizó con tratamiento continuo. En este estudio, la probabilidad de trombocitopenia al parecer aumenta significativamente a concentraciones totales de valproato de ≥110 mg/mL (mujeres) o ≥135 mg/mL (varones). Se debe valorar el beneficio terapéutico que puede acompañar a las dosis más altas contra la posibilidad de una mayor incidencia de reacciones adversas. Hiperamonemia: Se ha reportado hiperamonemia en asociación con el tratamiento con valproato y puede estar presente a pesar de las pruebas de función hepática con resultado normal. En pacientes que desarrollan letargo y vómito o cambios en el estado mental, debe considerarse la encefalopatía hiperamonémica y determinarse el nivel de amoniaco. También debe considerarse la hiperamonemia en pacientes que presentan hipotermia (ver Precauciones Generales - Hipotermia). Si el amoniaco aumenta, se debe suspender el tratamiento con valproato. Se deben iniciar intervenciones adecuadas para el tratamiento de hiperamonemia, y esos pacientes deben ser sometidos a investigación por trastornos subyacentes del ciclo de urea (ver Contraindicaciones y - Trastornos del ciclo de la urea y Precauciones Generales - Hiperamonemia y encefalopatía asociadas con el uso concomitante de topiramato). Las elevaciones asintomáticas de amoniaco son más comunes y, cuando se presentan requieren un monitoreo estrecho de los niveles plasmáticos de amoniaco. Si la elevación persiste, debe considerarse la suspensión del tratamiento con valproato. Hiperamonemia y encefalopatía asociadas con el uso concomitante de topiramato: La administración concomitante de topiramato y ácido valproico se ha asociado con hiperamonemia con o sin encefalopatía en pacientes que han tolerado cualquiera de los fármacos solos. Los síntomas clínicos de encefalopatía hiperamonémica a menudo incluyen alteraciones agudas en el nivel de conciencia y/o función cognitiva con letargo o vómito. La hipotermia también puede ser una manifestación de hiperamonemia (ver Precauciones Generales - Hipotermia). En la mayoría de los casos, los síntomas y signos disminuyeron con la suspensión de cualquiera de los dos fármacos. Este evento adverso no se debe a una interacción farmacocinética. Se desconoce si la monoterapia con topiramato está relacionada con hiperamonemia. Los pacientes con errores innatos en el metabolismo o menor actividad mitocondrial hepática pueden estar en mayor riesgo de hiperamonemia con o sin encefalopatía. Aunque no se ha estudiado, una interacción de topiramato y ácido valproico puede exacerbar los defectos existentes o desenmascarar deficiencias en personas sensibles (ver Contraindicaciones y precauciones generales - Trastornos del ciclo de la urea y Precauciones Generales - Hiperamonemia). Hipotermia: Se reportó hipotermia, definida como un descenso involuntario en la temperatura corporal central a < 35 °C (95 °F) relacionada con el tratamiento con valproato tanto con presencia y en ausencia de hiperamonemia. Esta reacción adversa también puede presentarse en pacientes que toman topiramato concomitante con valproato después de iniciar el tratamiento con topiramato o después de aumentar la dosis diaria de topiramato (ver Interacciones medicamentosas - Topiramato). Se debe considerar suspender el valproato en pacientes que desarrollan hipotermia, que puede manifestarse mediante una variedad de anomalías clínicas incluyendo letargo, confusión, coma y alteraciones significativas en otros sistemas de órganos principales como el aparato cardiovascular y respiratorio. El manejo clínico y evaluación deben incluir el análisis de niveles sanguíneos de amoniaco. General: Debido a los reportes de trombocitopenia (ver Precauciones Generales), la inhibición de la fase secundaria de agregación plaquetaria y parámetros de coagulación anormales (por ejemplo, fibrinógeno bajo), se recomienda realizar pruebas para determinar el número de plaquetas y coagulación antes de iniciar el tratamiento y en intervalos periódicos. Se recomienda monitorear los parámetros de plaquetas y coagulación de los pacientes que reciben valproato semisódico antes de una cirugía planeada. La evidencia de hemorragia, contusiones o un trastorno de hemostasias/coagulación es una indicación para reducir la dosis o suspender el tratamiento. Ya que valproato semisódico puede interactuar con los fármacos administrados concomitantemente que pueden inducir enzimas, se recomienda determinar periódicamente la concentración plasmática de valproato y los fármacos concomitantes durante la primera parte del tratamiento. Reacción de hipersensibilidad en múltiples órganos: En contadas ocasiones se han reportado reacciones de hipersensibilidad en múltiples órganos en asociación cercana temporal después de iniciar el tratamiento con valproato en pacientes adultos y pediátricos (mediana de tiempo hasta la detección 21 días; rango 1 a 40). A pesar de que el número de reportes es limitado, muchos de estos casos redundaron en hospitalización y se ha reportado por lo menos una muerte. Información para pacientes: Se debe advertir a los pacientes y cuidadores que el dolor abdominal, náusea, vómito y/o anorexia podrían ser síntomas de pancreatitis y, por lo tanto requieren evaluación médica inmediata. Los pacientes y cuidadores deben estar informados de los signos y síntomas asociados con encefalopatía hiperamonémica (ver Precauciones Generales - Hiperamonemia) y se les debe indicar que si se presenta cualquiera de estos síntomas lo informen al médico. Ya que los productos con valproato semisódico pueden producir depresión del SNC, especialmente cuando se combinan con otro depresivo del SNC (por ejemplo, alcohol), se debe recomendar a los pacientes que no participen en actividades peligrosas como manejar un automóvil u operar maquinaria peligrosa hasta que se sepa que el fármaco no les causa somnolencia. Debido a que el valproato sódico se ha asociado con ciertos tipos de defectos de nacimiento, se debe informar a las mujeres en edad fértil, que estén considerando el uso de valproato semisódico, los riesgos asociados con el uso de valproato semisódico durante el embarazo (véase Precauciones Generales).
Restricciones de su uso durante el embarzo y la lactancia: De acuerdo con los reportes publicados y sin publicar, el ácido valproico puede producir efectos teratogénicos, como defectos en el tubo neural (por ejemplo, espina bífida) en hijos de mujeres que reciben ácido valproico durante el embarazo. Hay datos que sugieren un aumento en la incidencia de malformaciones congénitas asociado con el uso de ácido valproico durante el embarazo cuando se comparó con otros fármacos antiepilépticos. Por consiguiente, el ácido valproico debe considerarse para mujeres en edad fértil sólo después de haber discutido exhaustivamente los riesgos con el paciente y de haberlos valorado contra los beneficios potenciales del tratamiento. Hay múltiples reportes en las publicaciones clínicas que indican que el uso de fármacos antiepilépticos durante el embarazo aumenta la incidencia de defectos de nacimiento. Por lo tanto, los fármacos antiepilépticos sólo deben administrarse a mujeres fértiles si han demostrado ser esenciales en el manejo de su enfermedad. Los datos que se describen a continuación se obtuvieron casi exclusivamente de mujeres que recibieron valproato para tratar la epilepsia. La incidencia de defectos del tubo neural en el feto puede aumentar en madres que reciben valproato durante el primer trimestre del embarazo. El organismo Centro para el Control de Enfermedades (CDC, por sus siglas en inglés) de Estados Unidos estimó que las mujeres expuestas al ácido valproico tienen un riesgo de 1 a 2% de tener hijos con espina bífida. Se han reportado otras anomalías congénitas (por ejemplo, defectos craneofaciales, malformaciones cardiovasculares y anomalías que abarcan diferentes sistemas corporales) compatibles e incompatibles con la vida. No hay datos suficientes para determinar la incidencia de estas anomalías congénitas. La incidencia más alta de anomalías congénitas en mujeres tratadas con fármacos antiepilépticos con trastornos de crisis convulsivas no puede considerarse como una relación de causa y efecto. Hay problemas metodológicos intrínsecos en la obtención de datos adecuados sobre la teratogenia de fármacos en humanos; los factores genéticos o la enfermedad epiléptica misma, pueden ser un factor de anomalías congénitas más importante que el tratamiento farmacológico. Existen reportes de retraso del desarrollo, autismo y/o trastorno del espectro de autismo en hijos de mujeres expuestas al ácido valproico durante el embarazo. Los pacientes que toman valproato pueden desarrollar alteraciones de la coagulación. Una paciente que tuvo fibrinógeno bajo cuando tomaba múltiples anticonvulsivos incluyendo valproato dio a luz un bebé con afibrinogenemia que posteriormente murió de hemorragia. Si se usa valproato durante el embarazo, se deben monitorear cuidadosamente los parámetros de coagulación. Se reportó insuficiencia hepática que causó la muerte de un recién nacido y de un lactante después del uso de valproato durante el embarazo. Las pruebas para detectar defectos del tubo neural y otros defectos usando los procedimientos actuales deben considerarse una parte de la rutina prenatal y en mujeres en edad fértil que reciben valproato. Los fármacos antiepilépticos no deben suspenderse abruptamente en pacientes a quienes se administra el fármaco para prevenir crisis convulsivas mayores debido a la fuerte posibilidad de precipitar estado epiléptico con hipoxia asociada y amenaza para la vida. En casos individuales en los que la severidad y frecuencia del trastorno de crisis convulsivas son tales que suspender la dosis del medicamento no representa una amenaza seria para el paciente, se puede considerar suspender el fármaco antes del embarazo y durante el mismo, aunque no es posible afirmar con seguridad que incluso las crisis menores no representan cierto peligro para el embrión o feto en desarrollo. Los estudios en animales han demostrado teratogenia inducida por valproato. En ratones, ratas, conejos y monos se ha observado aumento en la frecuencia de malformaciones, retraso del crecimiento intrauterino y muerte después de la exposición prenatal al valproato. Las malformaciones del sistema esquelético son las anomalías estructurales más comunes producidas en animales de experimentación, pero se han observado defectos de cierre del tubo neural en ratones expuestos a concentraciones plasmáticas maternas a valproato que exceden 230 mg/mL (2.3 veces el límite superior normal del rango terapéutico en humanos para epilepsia) durante los periodos sensibles del desarrollo embrionario. La administración de una dosis oral de 200 mg/kg/día o mayor (50% de la dosis diaria máxima en humanos o mayor en mg/m2) a ratas preñadas durante la organogénesis produjo malformaciones (esqueléticas, cardiacas y urogenitales) y retraso del crecimiento en las crías. Estas dosis produjeron niveles plasmáticos máximos maternos de valproato de aproximadamente 340 mg/mL o mayores (3.4 veces o más el límite superior del rango terapéutico en humanos). Se han reportado deficiencias conductuales en crías de ratas a las que se administró una dosis de 200 mg/kg/día durante la mayor parte del embarazo. Una dosis oral de 350 mg/kg/día (aproximadamente dos veces la dosis diaria máxima en humanos en mg/m2) produjo malformaciones esqueléticas y viscerales en conejos expuestos durante la organogénesis. Se observaron malformaciones esqueléticas, retraso del crecimiento y muerte en monos rhesus después de la administración de una dosis oral de 200 mg/kg/día (igual a la dosis diaria máxima en humanos en mg/m2) durante la organogénesis. Esta dosis produjo niveles plasmáticos máximos maternos de valproato de aproximadamente 280mg /mL (2.8 veces el límite superior del rango terapéutico en humanos). Lactancia: El valproato se excreta en la leche materna. Se ha reportado que las concentraciones en la leche materna son 1 a 10% de las concentraciones séricas. Se desconoce el efecto que esto tendría en un lactante alimentado con leche materna. Se debe considerar suspender la lactancia materna cuando se administra valproato semisódico a mujeres en etapa de lactancia.
Reacciones secundarias y adversas: Epilepsia: Crisis convulsivas parciales complejas (CPC): Con base en un estudio controlado con placebo de tratamiento complementario para crisis parciales complejas, valproato semisódico generalmente fue bien tolerado con la mayoría de los eventos adversos clasificados como de severidad leve a moderada. La intolerancia fue el motivo principal de suspensión en los pacientes tratados con valproato semisódico (6%), en comparación con el 1% de los pacientes tratados con placebo. La Tabla 1 enumera los eventos adversos derivados del tratamiento reportados por ≥ 5% de los pacientes tratados con valproato semisódico y para los cuales la incidencia fue mayor que en el grupo placebo, en el estudio controlado con placebo de tratamiento complementario de crisis convulsivas parciales complejas. Debido a que los pacientes también fueron tratados con otros fármacos antiepilépticos, en la mayoría de los casos no es posible determinar si los siguientes eventos adversos pueden atribuirse al valproato semisódico solo o a la combinación de valproato semisódico y otros fármacos antiepilépticos.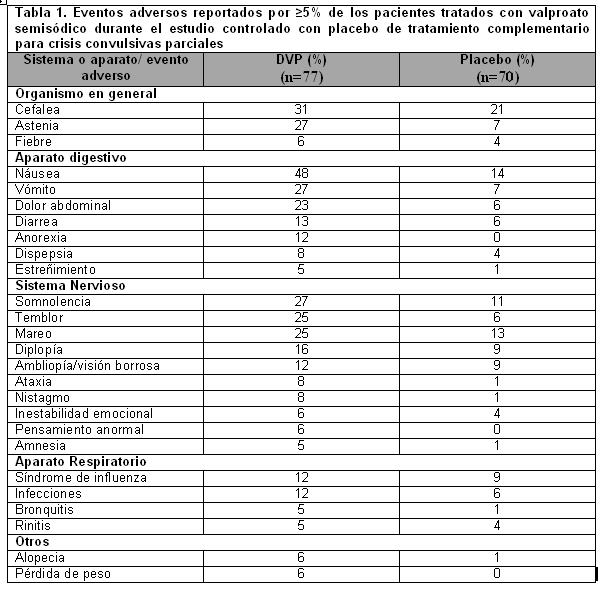
La Tabla 2 enumera los eventos adversos derivados del tratamiento reportados por ≥ 5% de los pacientes en el grupo de dosis alta de valproato semisódico y para los cuales la incidencia fue mayor que en el grupo de dosis baja, en un estudio controlado de monoterapia con valproato semisódico para crisis parciales complejas. Debido a que durante la primera parte del estudio se estaba ajustando otro fármaco antiepiléptico para suspenderlo, en muchos casos no es posible determinar si los siguientes eventos adversos pueden atribuirse a valproato semisódico solo o a la combinación de valproato semisódico y otros fármacos antiepilépticos.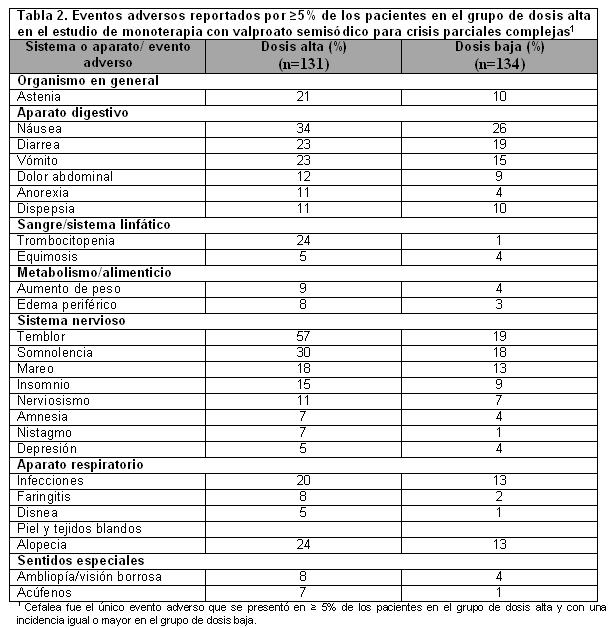
Los siguientes eventos adversos fueron reportados por más del 1% pero menos del 5% de los 358 pacientes tratados con valproato semisódico en los estudios controlados de crisis convulsivas parciales complejas: Organismo en general: dorsalgia, dolor precordial, malestar. Aparato cardiovascular: taquicardia, hipertensión, palpitación. Aparato digestivo: aumento del apetito, flatulencia, hematemesis, eructo, pancreatitis, absceso periodontal. Sangre y sistema linfático: petequias. Trastornos metabólicos y alimenticios: ALT elevada, AST elevada. Sistema musculoesquelético: mialgia, espasmos, artralgia, calambres en piernas, miastenia. Sistema nervioso: ansiedad, confusión, trastorno del habla, marcha anormal, parestesia, hipertonía, descoordinación, sueños anormales, trastorno de la personalidad. Aparato respiratorio: sinusitis, aumento de tos, neumonía, epistaxis. Piel y tejidos blandos: erupción cutánea, prurito, piel seca. Sentidos especiales: disgeusia, visión anormal, trastorno del oído, sordera, otitis media. Aparato urogenital: incontinencia urinaria, vaginitis, dismenorrea, amenorrea, frecuencia urinaria. Otras poblaciones de pacientes: A continuación se enumeran por sistema y aparato del organismo los eventos adversos que se han reportado con todas las formas farmacéuticas de valproato en estudios de epilepsia, reportes espontáneos y otras fuentes. Gastrointestinales: Los efectos reportados más comúnmente en el inicio del tratamiento son náusea, vómito e indigestión. Estos efectos generalmente son transitorios y rara vez es necesario suspender el tratamiento. Se han reportado diarrea, cólicos y estreñimiento. También se ha reportado anorexia con cierta pérdida de peso y aumento de apetito con aumento de peso. La administración de valproato semisódico con cubierta entérica puede resultar en la reducción de efectos secundarios gastrointestinales en algunos pacientes. Efectos en el SNC: Se han observado efectos sedantes en pacientes que reciben valproato solo pero se presentan con más frecuencia en pacientes que reciben tratamiento combinado. La sedación por lo regular disminuye al reducir el otro medicamento antiepiléptico. Se reportaron temblor (puede relacionarse con la dosis), alucinaciones, ataxia, cefalea, nistagmo, diplopía, asterixis, "manchas delante de los ojos", disartria, mareo, confusión, hipoestesia, vértigo, falta de coordinación y parkinsonismo con el uso de valproato. Se han presentado casos contados de coma en pacientes que reciben valproato solo o en combinación con fenobarbital. En contados casos se ha desarrollado encefalopatía con o sin fiebre poco después de la introducción de monoterapia con valproato sin evidencia de disfunción