VELMETIA
LIOMONT
Denominación genérica: Metformina, Sitagliptina
Forma farmacéutica y formulación: Comprimidos.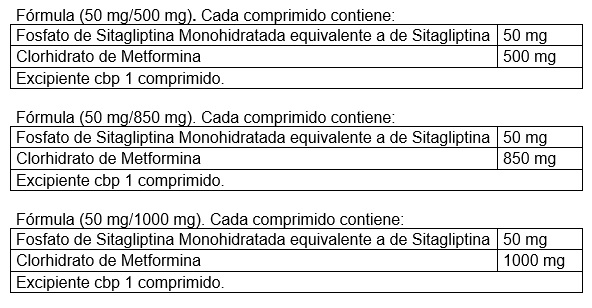
Indicaciones terapéuticas: VELMETIA está indicado como tratamiento inicial en pacientes con diabetes mellitus tipo 2 para mejorar el control glucémico cuando la dieta y el ejercicio no brindan un control adecuado de la glucemia. VELMETIA está indicado como complemento de la dieta y el ejercicio para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2 que no están controlados suficientemente con metformina o sitagliptina solas o en los que ya están siendo tratados con sitagliptina y metformina. VELMETIA está indicado como parte del tratamiento combinado triple con una sulfonilurea como complemento de la dieta y el ejercicio, en pacientes con diabetes mellitus tipo 2 inadecuadamente controlados con cualquiera de dos de los tres medicamentos: metformina, sitagliptina o una sulfonilurea. VELMETIA está indicado como parte del tratamiento combinado triple con un agonista del PPARg (por ejemplo, tiazolidinedionas) como complemento de la dieta y el ejercicio, en pacientes con diabetes mellitus tipo 2 inadecuadamente controlados con cualquiera de dos de los tres medicamentos: metformina, sitagliptina o un agonista del PPARg. VELMETIA está indicado en pacientes con diabetes mellitus tipo 2 como complemento a la dieta y el ejercicio para mejorar el control de la glucemia en combinación con insulina.
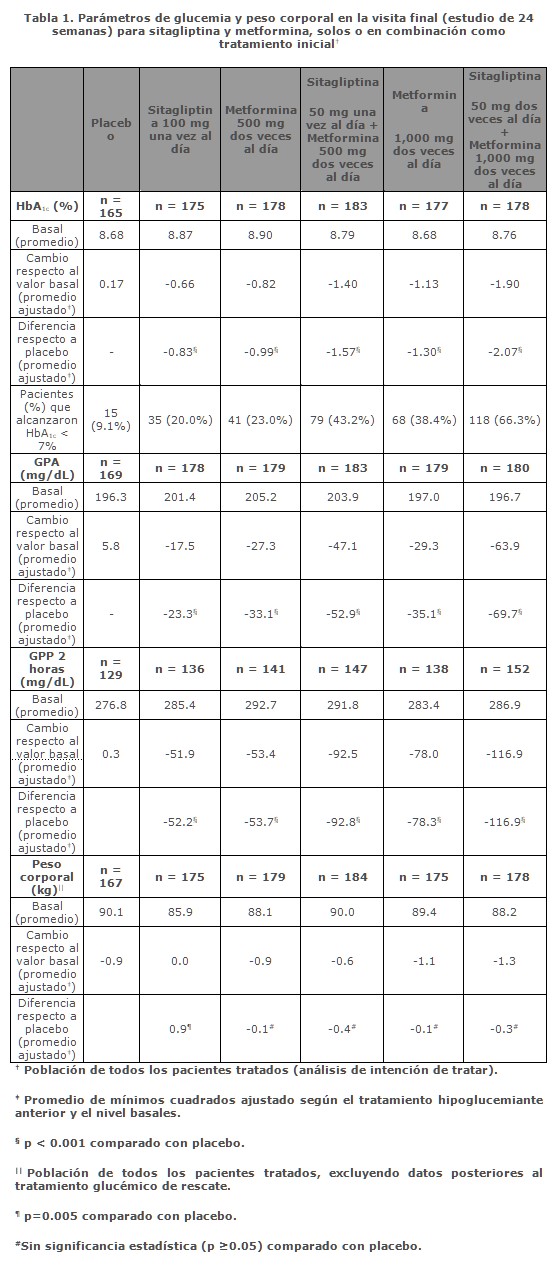
Farmacocinética y farmacodinamia: Mecanismo de acción: VELMETIA combina dos agentes antihiperglucemiantes con mecanismos de acción complementarios para mejorar el control de la glucemia en pacientes con diabetes tipo 2: Fosfato de sitagliptina, que es un inhibidor de la dipeptidil peptidasa 4 (DPP-4), y clorhidrato de metformina, que es una biguanida. Las tabletas de VELMETIA contienen sitagliptina y una formulación de metformina de liberación inmediata. Fosfato de sitagliptina: El fosfato de sitagliptina es un miembro de una clase de agentes hipoglucemiantes llamados inhibidores de la dipeptidil peptidasa 4 (DPP-4), que mejoran el control glucémico en pacientes con diabetes tipo 2 aumentando las concentraciones de las hormonas incretinas activas. Estas hormonas, que incluyen el péptido-1 similar al glucagón (GLP-1) y el péptido insulinotrópico dependiente de la glucosa (GIP), son liberadas por el intestino durante todo el día y sus concentraciones aumentan en respuesta a la ingestión de alimentos. Las incretinas forman parte de un sistema endógeno que participa en la regulación fisiológica de la homeostasis de la glucosa. Cuando la glucemia está normal o elevada, el GLP-1 y el GIP aumentan la síntesis y la liberación de insulina por las células beta del páncreas a través de vías de señalización intracelulares en las que interviene el monofosfato de adenosina cíclico (AMPc). Se ha demostrado que el tratamiento con inhibidores del GLP-1 o de la DPP-4 en modelos animales de diabetes tipo 2 mejora la capacidad de respuesta de las células beta a la glucosa y estimula la biosíntesis y la liberación de insulina. Con concentraciones mayores de insulina, aumenta la captación tisular de glucosa. Además, el GLP-1 disminuye la secreción de glucagón por las células alfa del páncreas. Las concentraciones disminuidas de glucagón y el aumento de las concentraciones de insulina hacen que disminuya la producción hepática de glucosa, lo cual da por resultado la disminución de la concentración de glucosa en la sangre. Los efectos del GLP-I y del GIP son dependientes de la glucosa, de tal manera que cuando la glucemia está baja no se observa la estimulación de la liberación de insulina ni la inhibición de la secreción de glucagón por el GLP-1. La estimulación de liberación de insulina, tanto por GLP-1 como por GIP, se potencializa cuando los niveles de glucosa están por arriba de lo normal. Más aún, GLP-1 no afecta la respuesta normal del glucagón a la hipoglucemia. La actividad de GLP-1 y de GIP está limitada por la enzima DPP-4, que hidroliza rápidamente las incretinas y las transforma en productos inactivos. La sitagliptina impide esa hidrólisis de las incretinas por la DPP-4, por lo que aumentan las concentraciones plasmáticas de las formas activas del GLP-1 y del GIP. Como consecuencia, la sitagliptina aumenta la liberación de insulina y disminuye la concentración de glucagón de manera dependiente de la glucosa. En los pacientes con diabetes tipo 2 con hiperglucemia, esos cambios en las concentraciones de insulina y de glucagón hacen que disminuyan las concentraciones de hemoglobina A1c (HbA1c) y la glucemia en ayunas y post-prandial. El mecanismo dependiente de glucosa de sitagliptina es diferente al de sulfonilureas, ya que éstas incrementan la secreción de insulina aun cuando los niveles de glucosa estén bajos, lo que puede causar hipoglucemia en pacientes con diabetes tipo 2 y en sujetos normales. Aunque la sitagliptina es un inhibidor potente y sumamente selectivo de la enzima DPP-4, no inhibe las enzimas estrechamente relacionadas DPP-8 y DPP-9, a concentraciones terapéuticas. Clorhidrato de metformina: La metformina es un agente hipoglucemiante que mejora la tolerancia a la glucosa en pacientes con diabetes tipo 2 disminuyendo tanto la glucosa plasmática basal como la post-prandial. Sus mecanismos de acción farmacológicos son diferentes de los de otras clases de hipoglucemiantes orales. La metformina disminuye la producción hepática y la absorción intestinal de glucosa, y mejora la sensibilidad a la insulina aumentando la captación y la utilización periférica de glucosa. A diferencia de las sulfonilureas, la metformina no produce hipoglucemia ni en los pacientes con diabetes tipo 2 ni en los sujetos normales (excepto en circunstancias especiales; véase Precauciones generales, Clorhidrato de metformina), y no causa hiperinsulinemia. El tratamiento con metformina no modifica la secreción de insulina, mientras que puede disminuir los niveles de insulina en ayunas y la respuesta de la insulina plasmática durante todo el día. Farmacocinética: VELMETIA: Los resultados de un estudio de bioequivalencia definitivo en sujetos sanos demostraron que los comprimidos de VELMETIA (Sitagliptina/Clorhidrato de metformina) con 50 mg/500 mg y con 50 mg/1000 mg son bioequivalentes a la coadministración de esas mismas dosis de fosfato de sitagliptina y clorhidrato de metformina en comprimidos individuales. Debido a que se demostró bioequivalencia a las dosis más baja y más alta de tabletas combinadas, se confirió bioequivalencia a la tableta de combinación fija (FDC) de 50 mg/850 mg (sitagliptina/metformina). Absorción: VELMETIA: Posterior a la ingesta de las tabletas de VELMETIA con un desayuno alto en grasas, el grado total de exposición (ABC) y la tasa de absorción (Cmáx y Tmáx) para sitagliptina no se alteraron en comparación con el estado de ayuno. Posterior a la ingesta de tabletas de VELMETIA con un desayuno alto en grasas, el ABC y Cmáx de metformina disminuyeron en 6% y 28% respectivamente, y el Tmáx ocurrió aproximadamente 1.5 horas más tarde, en relación con el estado de ayuno. Fosfato de sitagliptina: La biodisponibilidad absoluta de la sitagliptina es de 87% aproximadamente. La coadministración de una comida alta en grasas con fosfato de sitagliptina no tuvo ningún efecto sobre la farmacocinética de la sitagliptina. Clorhidrato de metformina: La biodisponibilidad absoluta de un comprimido de 500 mg de clorhidrato de metformina administrado en ayunas es de 50-60% aproximadamente. Los estudios con dosis orales únicas de tabletas de clorhidrato de metformina de 500 a 1,500 mg y de 850 a 2,550 mg indican que el efecto no es proporcional al incremento de las dosis, lo cual se debe a una disminución de la absorción y no a una alteración en la eliminación. La presencia de alimentos disminuye y retarda ligeramente la absorción de la metformina, como muestran una disminución de 40% aproximadamente en su concentración plasmática máxima (Cmáx), una disminución de 25% del ABC de concentración plasmática y un aumento de 35 minutos del tiempo para alcanzar la concentración máxima plasmática (Tmáx) tras la administración de un solo comprimido de 850 mg de metformina con alimentos, en comparación con la administración de esa misma dosis en ayunas. Se desconoce la importancia clínica de estas disminuciones. Los alimentos bajos y altos en grasa incrementaron la exposición sistémica (medido por el ABC) de las tabletas de Glumetza alrededor de un 38% y 73%, respectivamente, en relación con el ayuno. Ambos alimentos prolongaron el Tmáx de metformina en aproximadamente 3 horas, pero la Cmáx no fue afectada. Distribución: Fosfato de sitagliptina: El volumen de distribución promedio en estado de equilibrio tras una dosis única intravenosa de sitagliptina a personas sanas es de 198 litros aproximadamente. La porción de sitagliptina que se une reversiblemente a las proteínas plasmáticas es baja (38%). Clorhidrato de metformina: El volumen aparente de distribución (V/F) de la metformina tras la administración única de tabletas con 850 mg de clorhidrato de metformina fue en promedio de 654 ± 358 litros. A diferencia de las sulfonilureas, que se unen en más de 90% a las proteínas plasmáticas, la metformina se une escasamente a éstas. Parte de la metformina penetra en los eritrocitos, muy probablemente en función del tiempo. A las dosis clínicas y horarios de administración usuales de las tabletas de clorhidrato de metformina, las concentraciones plasmáticas máximas de metformina en el estado de equilibrio se alcanzan en 24 a 48 horas y son generalmente menores de 1 mcg/mL. Durante los estudios clínicos controlados de metformina, sus concentraciones plasmáticas máximas no fueron mayores de 5 mcg/mL, aun con las dosis máximas. Metabolismo:Fosfato de sitagliptina: La sitagliptina es eliminada princi-palmente sin cambio en la orina, y su transformación metabólica es una vía menor. Aproximadamente 79% es excretada sin cambio en la orina. Después de una dosis oral de sitagliptina marcada con 14C, aproximadamente 16% de la radiactividad fue excretada como metabolitos de la sitagliptina. Se detectaron sólo trazas de seis metabolitos, y no se prevé que éstos contribuyan a la actividad inhibidora de la sitagliptina sobre la DPP-4 plasmática. Los estudios in vitro indicaron que la principal enzima causante del metabolismo limitado de la sitagliptina fue CYP3A4, secundada por CYP2C8. Clorhidrato de metformina: Los estudios con dosis únicas por vía intravenosa en sujetos normales demostraron que la metformina es excretada sin cambio en la orina y no es metabolizada en el hígado (no se ha identificado ningún metabolito en los seres humanos) ni excretada con la bilis. Eliminación: Fosfato de sitagliptina: Tras la administración de una dosis oral de [14C]sitagliptina a sujetos sanos, aproximadamente 100% de la radiactividad fue eliminada en las heces (13%) o la orina (87%) durante la semana siguiente. Después de una dosis oral de 100 mg, la t½ terminal aparente de la sitagliptina fue de 12.4 horas aproximadamente, y su depuración renal fue de unos 350 mL/min. La eliminación de la sitagliptina ocurre principalmente por excreción renal, por secreción tubular activa. La sitagliptina es un sustrato para el transportador 3 de aniones orgánicos humanos (hOAT-3), el cual podría intervenir en su eliminación renal. No se ha determinado la importancia clínica del hOAT-3 en el transporte de la sitagliptina. Ésta es también un sustrato de la p-glucoproteína, que también puede mediar en su eliminación renal. Sin embargo, la ciclosporina, que es un inhibidor de la p-glucoproteína, no disminuyó la depuración renal de la sitagliptina. Clorhidrato de metformina: La depuración renal de la metformina es aproximadamente 3.5 veces mayor que la de la creatinina, lo cual indica que la secreción tubular es la principal vía de eliminación de la metformina. Tras la administración por vía oral, aproximadamente 90% del medicamento absorbido es eliminado por la vía renal en las primeras 24 horas, con una vida media de eliminación plasmática de 6.2 horas aproximadamente. En la sangre, la vida media de eliminación es de unas 17.6 horas aproximadamente, lo cual sugiere que la masa eritrocitaria puede ser un compartimiento de distribución del fármaco. Características de los pacientes: Diabetes tipo 2: Fosfato de sitagliptina: La farmacocinética de la sitagliptina es generalmente similar en los pacientes con diabetes tipo 2 y en los sujetos sanos. Clorhidrato de metformina: Si la función renal es normal, no hay ninguna diferencia en la farmacocinética de la metformina administrada en dosis únicas o múltiples entre los pacientes con diabetes tipo 2 y los sujetos sanos, y a las dosis clínicas usuales no ocurre ninguna acumulación del medicamento en ninguno de esos dos grupos. Insuficiencia renal: Fosfato de sitagliptina: En comparación con los sujetos con función renal normal, el ABC plasmático de la sitagliptina aumentó aproximadamente al doble en los pacientes con insuficiencia renal moderada con una Tasa de Filtración Glomerular estimada (TFGe) de 30 a < 45 mL/min/1.73 m² y aproximadamente al cuádruple en los pacientes con insuficiencia renal grave (TFGe < 30 mL/min/1.73 m² incluyendo a los pacientes con nefropatía terminal bajo tratamiento con hemodiálisis. Clorhidrato de metformina: En los pacientes con disminución de la función renal aumenta la vida media plasmática y sanguínea de la metformina y disminuye su depuración renal (Véase Contraindicaciones y Precauciones generales). Insuficiencia hepática: Fosfato de sitagliptina: En los pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), tras la administración de una dosis única de 100 mg de fosfato de sitagliptina los promedios de ABC y de Cmáx. de sitagliptina fueron aproximadamente 21% y 13% mayores, respectivamente, que en los testigos sanos. Esas diferencias no se consideran clínicamente importantes. No se tiene ninguna experiencia clínica en pacientes con insuficiencia hepática grave (puntuación de Child-Pugh mayor de 9). Sin embargo, como la sitagliptina se elimina principalmente por vía renal, no se prevé que la insuficiencia hepática grave afecte la farmacocinética de la sitagliptina. Clorhidrato de metformina: No se han hecho estudios sobre la farmacocinética de la metformina en pacientes con insuficiencia hepática. Sexo: Fosfato de sitagliptina: Basándose en un análisis compuesto de los datos farmacocinéticos de la Fase I y en un análisis farmacocinético de población de los datos de las Fases I y II, el sexo no tuvo ningún efecto de importancia clínica sobre la farmacocinética de la sitagliptina. Clorhidrato de metformina: Los parámetros farmacocinéticos de la metformina no difirieron significativamente entre las personas normales y los pacientes con diabetes tipo 2 cuando se analizaron según el sexo, y en los estudios clínicos controlados en pacientes con diabetes tipo 2 el efecto hipoglucemiante de la metformina fue similar en los hombres y en las mujeres. Pacientes de edad avanzada: Fosfato de sitagliptina: Según un análisis farmacocinético de población de los datos de las Fases I y II, la edad no tuvo un efecto de importancia clínica sobre la farmacocinética de la sitagliptina. Las personas de edad avanzada (65 a 80 años) tuvieron concentraciones plasmáticas de sitagliptina aproximadamente 19% mayores que en sujetos de menor edad. Clorhidrato de metformina: Datos limitados de estudios farmacocinéticos controlados de metformina en personas de edad avanzada sanas sugieren que en éstas está disminuida la depuración plasmática total de la metformina y aumentadas su vida media y su Cmáx en comparación con personas jóvenes sanas. Según estos datos, el cambio en la farmacocinética de la metformina al aumentar la edad es debido principalmente al cambio en la función renal (véase la información para prescribir de Glucophage†). Niños: No se ha hecho ningún estudio con VELMETIA en niños. Raza: Fosfato de sitagliptina: Basándose en un análisis compuesto de los datos farmacocinéticos de la Fase I y en un análisis farmacocinético de población de los datos de las Fases I y II que incluyeron personas de razas blanca y negra, Hispanoamericanos, Asiáticos y de otros grupos raciales, la raza no tuvo ningún efecto de importancia clínica sobre la farmacocinética de la sitagliptina. Clorhidrato de metformina: No se han hecho estudios de los parámetros farmacocinéticos de la metformina según la raza. En estudios clínicos controlados de metformina en pacientes con diabetes tipo 2, su efecto hipoglucemiante fue similar en blancos (n = 249), negros (n = 51) e Hispanoamericanos (n = 24). Índice de masa corporal: Fosfato de sitagliptina: Basándose en un análisis compuesto de los datos farmacocinéticos de la Fase I y en un análisis farmacocinético de población de los datos de las Fases I y II, el índice de masa corporal no tuvo ningún efecto de importancia clínica sobre la farmacocinética de la sitagliptina. Farmacodinamia: Fosfato de sitagliptina: General: En pacientes con diabetes tipo 2, la administración de dosis orales únicas de sitagliptina inhibe la actividad de la enzima DPP-4 durante 24 horas, lo cual aumenta al doble o al triple las concentraciones circulantes de GLP-1 y GIP activos, aumenta las concentraciones plasmáticas de insulina y de péptido C, y disminuye las concentraciones de glucagón, la glucemia en ayunas y el margen de variación de la glucemia después de la ingestión de glucosa o de una comida. En los estudios clínicos Fase III de 18 y 24 semanas de duración, el tratamiento con 100 mg diarios de sitagliptina en pacientes con diabetes tipo 2 mejoró significativamente el funcionamiento de las células beta evaluado mediante varios marcadores, que incluyeron la Evaluación Modelo ß de Homeostasis (HOMA-ß, por sus siglas en inglés), la relación proinsulina/insulina y las mediciones de la sensibilidad de las células beta según las muestras múltiples de la prueba de tolerancia a la ingestión de alimentos. En estudios de Fase II, la eficacia sobre la glucemia de la dosificación de 50 mg de sitagliptina dos veces al día fue similar a la de 100 mg una vez al día. En un estudio doble ciego de dos días, cruzado, de cuatro periodos, con distribución al azar y controlado, de doble enmascaramiento en adultos sanos, se compararon los efectos de la coadministración de sitagliptina y metformina sobre las concentraciones de GLP-1 plasmático activo y total y la glucemia post-prandiales con los de la administración de sitagliptina sola, metformina sola o placebo, cada uno administrado por dos días. El aumento del promedio ponderado de las concentraciones de GLP-1 activo cuatro horas después de la comida fue aproximadamente dos veces mayor con la sitagliptina sola o la metformina sola que con el placebo. La coadministración de sitagliptina y metformina tuvo un efecto aditivo sobre las concentraciones de GLP-1 activo, que aumentaron unas cuatro veces más que con el placebo. La sitagliptina sola también aumentó sólo las concentraciones de GLP-1 activo, debido muy probablemente a la inhibición de la DPP-4, mientras que la metformina sola aumentó en grado similar las concentraciones de GLP-1 activo y total, lo cual refleja un mecanismo diferente para el aumento del GLP-1 activo, debido principalmente a un aumento del GLP-1 total. Estos datos son consistentes con diferentes mecanismos de acción para incrementar las concentraciones de GlP-1 activo. Los resultados de ese estudio también demostraron que la sitagliptina, pero no la metformina, aumenta las concentraciones de GIP activo. En estudios con sujetos sanos la sitagliptina no disminuyó la glucemia ni provocó hipoglucemia, lo cual sugiere que sus acciones insulinotrópica y supresora del glucagón son dependientes de la glucosa. Efectos sobre la presión arterial: En un estudio cruzado, controlado con placebo y con distribución al azar en pacientes hipertensos tratados con uno o más antihipertensivos (que incluyeron inhibidores de la enzima convertidora de angiotensina, antagonistas de la angiotensina II, bloqueadores de canales de calcio, bloqueadores beta y diuréticos), la coadministración con sitagliptina fue generalmente bien tolerada. En esos pacientes, la sitagliptina tuvo un modesto efecto reductor de la presión arterial; 100 mg diarios disminuyeron unos 2 mm Hg el promedio de presión sistólica ambulatoria en comparación con el placebo. No se han observado disminuciones en personas normotensas. Electrofisiología cardiaca: En un estudio cruzado, controlado con placebo y con distribución al azar, se administró a 79 sujetos sanos una dosis oral única de 100 mg de sitagliptina, de 800 mg de sitagliptina (ocho veces mayor que la dosis recomendada), y placebo. La dosis recomendada de 100 mg no tuvo ningún efecto sobre el intervalo QTc obtenido durante la concentración plasmática máxima o en cualquier otro momento del estudio. Con la dosis de 800 mg, el aumento máximo del promedio de cambio del QTc inicial corregido respecto al placebo fue de sólo 8.0 mseg tres horas después de la administración. Este pequeño aumento no se consideró clínicamente importante. Con la dosis de 800 mg, las concentraciones plasmáticas máximas de sitagliptina fueron aproximadamente once veces mayores que las producidas por la dosis de 100 mg. En los pacientes con diabetes tipo 2 que recibieron sitagliptina a dosis de 100 mg diarios (N = 81) o de 200 mg diarios (N = 63) no hubo cambios importantes del intervalo QTc en los electrocardiogramas obtenidos en el momento de la concentración plasmática máxima esperada. Estudios clínicos: Los estudios clínicos con la coadministración de sitagliptina y metformina demostraron mejorías significativas en el control glucémico en pacientes con diabetes tipo 2. Ninguno de los estudios clínicos de eficacia descritos abajo se realizaron con VELMETIA comprimidos; sin embargo, se ha demostrado la bioequivalencia de VELMETIA comprimidos con la coadministración de sitagliptina y clorhidrato de metformina de liberación inmediata para todas las concentraciones de las tabletas, respectivamente. En un estudio comparativo, la administración una vez al día de metfomina de liberación prolongada ha tenido una eficacia similar a la de la formulación de metformina de liberación inmediata dos veces al día, en todas las medidas de control de la glucemia. Sitagliptina y metformina como tratamiento inicial en pacientes con diabetes tipo 2: Un total de 1,091 pacientes con diabetes tipo 2 y control inadecuado de la glucemia con dieta y ejercicio, participaron en un estudio factorial de 24 semanas, con distribución al azar, doble ciego, controlado con placebo, diseñado para evaluar la eficacia y seguridad del tratamiento inicial con la combinación de sitagliptina y metformina. Se distribuyó al azar a un número aproximadamente equivalente de pacientes para recibir tratamiento inicial con placebo; sitagliptina 100 mg una vez al día; metformina 500 o 1,000 mg dos veces al día; o sitagliptina 50 mg dos veces al día combinada con metformina 500 o 1,000 mg dos veces al día. El tratamiento inicial con la combinación de sitagliptina y metformina produjo mejorías significativas en HbA1C, en glucosa plasmática en ayunas (GPA) y en glucosa plasmática post-pandrial (GPP) a las 2 horas en comparación con placebo, con metformnina sola y con sitagliptina sola (p < 0.001; Tabla 1, Figura 1). A las 3 semanas (el primer punto de evaluación después de iniciar el tratamiento), se logró una mejoría en GPA cercana a la máxima reducción de GPA y se mantuvo durante las 24 semanas del estudio. Las mediciones de la función de las células beta, HOMA-ß y la relación proinsulina a insulina también mostraron más mejoría con la coadministración de sitagliptina y metformina en comparación con la monoterapia de ambos medicamentos. En general, los efectos en los lípidos fueron neutrales. La disminución en el peso corporal en el grupo de sitagliptina y metformina fue similar al de los grupos de metformina sola o de placebo.Las reducciones promedio respecto al inicio en HbA1C comparado con placebo fueron generalmente mayores en los pacientes que al inicio tenían valores más altos de HbA1C. La mejoría en HbA1c fue generalmente consistente entre los sub-grupos definidos por sexo, edad, raza o IMC inicial. Las reducciones promedio respecto al inicio en HbA1c de los pacientes que no estaban bajo tratamiento hipoglucemiante al inicio del estudio fueron: sitagliptina 100 mg una vez al día, -1.06%; metformina 500 mg dos veces al día, -1.09%; metformina 1,000 mg dos veces al día, -1.24%; sitagliptina 50 mg dos veces al día más metformina 500 mg dos veces al día, -1.59%; sitagliptina 50 mg dos veces al día más metformina 1,000 mg dos veces al día, -1.94% y; para quienes recibieron placebo, -0.17%.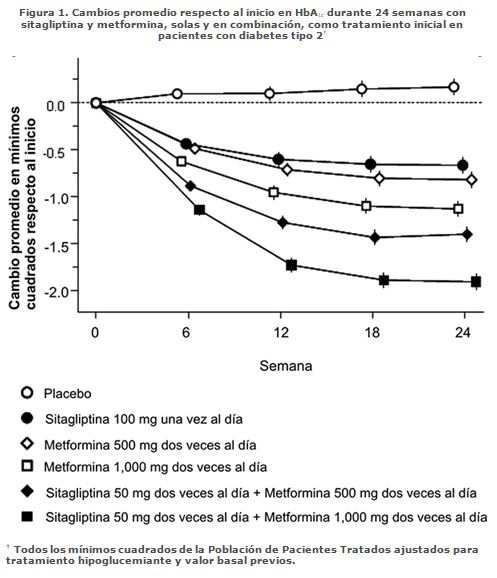
Además, este estudio incluyó pacientes (n = 117) con hiperglucemia más severa (HbA1C > 11%) o glucosa sanguínea > 280 mg/dL) que fueron tratados en forma abierta con sitagliptina 50 mg más metformina 1,000 mg dos veces al día. En este grupo de pacientes, el valor inicial de HbA1C era 11.15%, GPA era 314.4 mg/dL y GPP a las 2 horas era 441.0 mg/dL. Después de 24 semanas se observaron disminuciones de -2.94% para HbA1C, -126.7 mg/dL para GPA y -207.9 mg/dL para GPP a las dos horas. En esta cohorte abierta se observó un modesto incremento del peso corporal de 1.3 kg a las 24 semanas. Adición de sitagliptina en pacientes controlados inadecuadamente con metformina sola: La seguridad y la eficacia de la combinación de sitagliptina y metformina se evaluó en dos estudios clínicos doble ciego, controlados con placebo en pacientes con diabetes mellitus tipo 2. En ambos estudios se distribuyó al azar a pacientes en los que dosis estables de 1,500 mg o más de metformina no controlaban suficientemente la glucemia, para recibir 100 mg diarios de sitagliptina o placebo añadidos a su tratamiento con metformina. En un estudio, 701 pacientes recibieron 100 mg de sitagliptina o placebo una vez al día durante 24 semanas. En comparación con el placebo, la adición de sitagliptina al tratamiento con metformina mejoró significativamente la concentración de HbA1c (-0.65%), la GPA (-25.4 mg/dL) y la GPP a las dos horas (-50.6 mg/dL) (Figura 2 y Tabla 2). En esa mejoría de la HbA1c respecto a placebo, no influyeron su concentración basal, el tratamiento hipoglucemiante previo, el sexo, la edad, el índice de masa corporal basal, el tiempo transcurrido desde que se diagnosticó la diabetes, la presencia de síndrome metabólico, ni los índices estándares de resistencia a la insulina (HOMA-IR) o de secreción de insulina (HOMA-ß). Comparado con los pacientes que tomaron placebo, los pacientes que recibieron sitagliptina presentaron ligeras disminuciones en colesterol total, colesterol no HDL y en los triglicéridos. En los dos grupos de tratamiento se observó una disminución similar del peso corporal.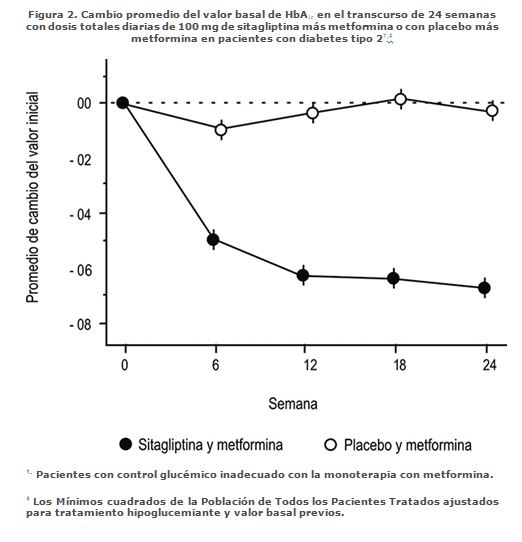
Pacientes con insuficiente control de la glucemia bajo monoterapia con metformina. ‡Todos los pacientes tratados. Promedios de los cuadrados mínimos ajustados según el tratamiento hipoglucemiante anterior y el valor inicial.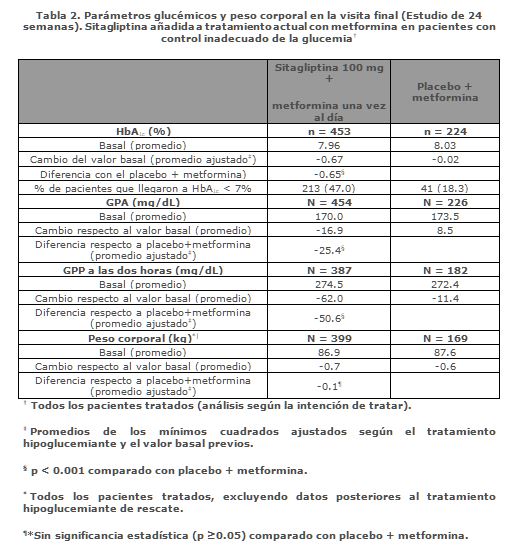
En otro estudio se midió la glucemia durante las 24 horas del día. Veintiocho pacientes recibieron 50 mg de sitagliptina o placebo dos veces al día durante cuatro semanas además de su tratamiento con metformina dos veces al día. Al cabo de esas cuatro semanas se determinó la diferencia en la eficacia reductora de la glucemia según el promedio ponderado de la glucemia en 24 horas, basándose en la toma de múltiples muestras de sangre, incluyendo las obtenidas antes y después de las comidas y por la noche. En comparación con el placebo más metformina, la coadministración de 50 mg de sitagliptina dos veces al día con la metformina disminuyó significativamente (-32.8 mg/dL) el promedio ponderado de la glucemia en 24 horas, disminuyó considerablemente la glucemia en ayunas, y causó menores variaciones de la glucemia después de las tres comidas del día (Figura 3). En las mediciones de la glucemia hechas por los pacientes, el tratamiento con sitagliptina más metformina también disminuyó significativamente en comparación con el placebo más metformina el promedio de glucemia en ayunas (-20.3 mg/dL), el promedio de siete mediciones de la glucemia (-28.0 mg/dL) y la glucemia postprandial a las dos horas (-36.6 mg/dL).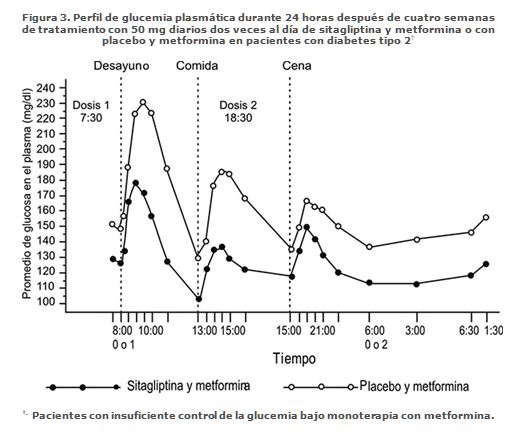
Tratamiento de adición de sitagliptina a pacientes controlados inadecuadamente con la combinación de glimepirida y metformina: Un total de 441 pacientes con diabetes tipo 2 participaron en un estudio de 24 semanas, con distribución al azar, doble ciego, controlado con placebo, que se diseñó para evaluar la eficacia de sitagliptina 100 mg una vez al día en combinación con glimepirida (sola o en combinación con metformina). En este estudio 220 pacientes estuvieron en la combinación de glimepirida (≥4 mg diarios) y metformina (≥1500 mg diarios). Los resultados de los puntos finales de glucemia, incluyendo HbA1c y glucosa en ayunas, se describen a continuación. La combinación de sitagliptina, glimepirida y metformina produjo, respecto al nivel basal, reducciones significativas en HbA1c (-0.89%) y GPA (-20.7 mg/dL) en comparación con placebo (véase Tabla 3). Las reducciones promedio respecto al nivel basal en HbA1c comparado con placebo en general fueron mayores en quienes tenían valores mayores de HbA1C al inicio. Los pacientes tratados con sitagliptina tuvieron un modesto incremento en el peso corporal respecto a quienes recibieron placebo.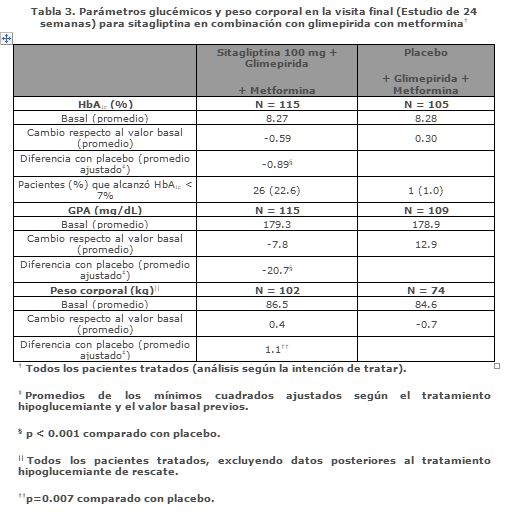
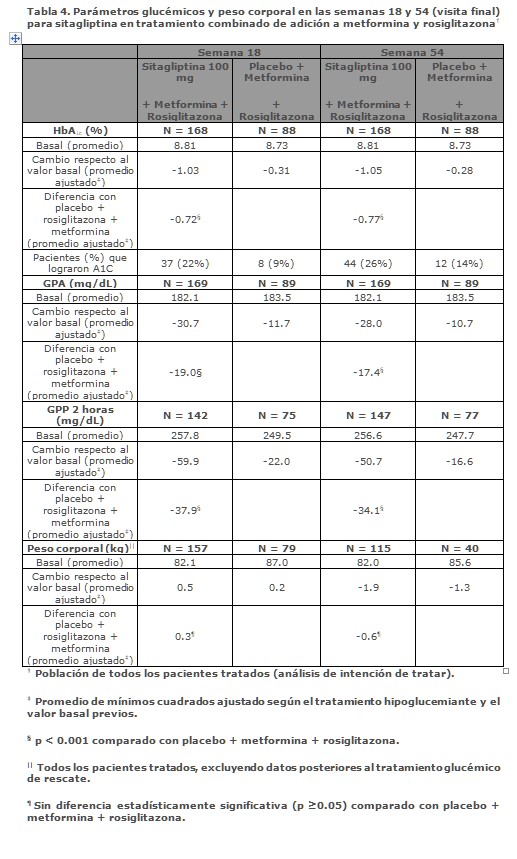
Tratamiento combinado de adición en pacientes controlados inadecuadamente con metformina más rosiglitazona: Un total de 262 pacientes con diabetes tipo 2 participaron en un estudio controlado con placebo, de 54 semanas, doble ciego, con distribución al azar diseñado para evaluar la eficacia de sitagliptina en combinación con metformina y rosiglitazona. Pacientes con control glucémico inadecuado con un esquema estable de metformina (≥1,500 mg diarios) y rosiglitazona (≥4 mg diarios) fueron distribuidos al azar a la adición de 100 mg de sitagliptina o de placebo administrados una vez al día. Los parámetros glucémicos fueron evaluados en el primer punto en el tiempo de la Semana 18 y a la Semana 54. En combinación con metformina y rosiglitazona, sitagliptina brindó mejorías significativas en HbA1c, GPA y GPP de 2 horas en comparación a placebo con metformina y rosiglitazona (Tabla 4, Figura 4) a la Semana 18, con mejorías sostenidas hasta el final del estudio. Los efectos en los lípidos fueron generalmente neutrales. No hubo diferencias significativas entre sitagliptina y placebo en el cambio en el peso corporal.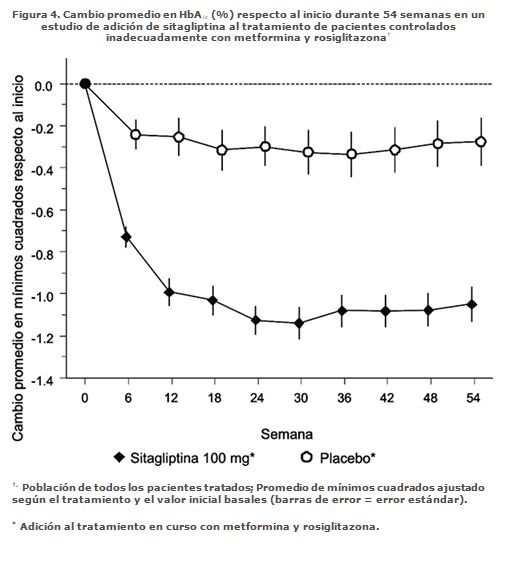
Tratamiento combinado de adición de sitagliptina en pacientes controlados inadecuadamente con la combinación de metformina e insulina: Un total de 641 pacientes con diabetes tipo 2 participaron en un estudio controlado con placebo, doble ciego, con distribución al azar, de 24 semanas diseñado para evaluar la eficacia de sitagliptina 100 mg una vez al día en combinación con una dosis estable de insulina. Aproximadamente 75% de los pacientes estaban también tomando metformina. Pacientes con insulina premezclada, de acción larga o de acción intermedia (con o sin metformina), fueron distribuidos al azar para que se les añadiera sitagliptina 100 mg o placebo. Los puntos finales glucémicos incluyeron HbA1c, glucosa en ayunas y glucosa post-prandial de 2 horas. La combinación de sitagliptina, metformina e insulina proporcionó mejorías significativas en HbA1c, glucosa en ayunas y glucosa post-prandial de 2 horas comparado con placebo (Tabla 5). La mejoría en HbA1c comparado con placebo fue generalmente consistente entre los subgrupos definidos por sexo, edad, raza, IMC inicial, tiempo desde el diagnóstico de diabetes, presencia de síndrome metabólico, e índices estándar de resistencia la insulina (HOMA-IR) y de secreción de insulina (HOMA-ß). No hubo diferencias importantes en el peso corporal respecto al inicio entre los grupos.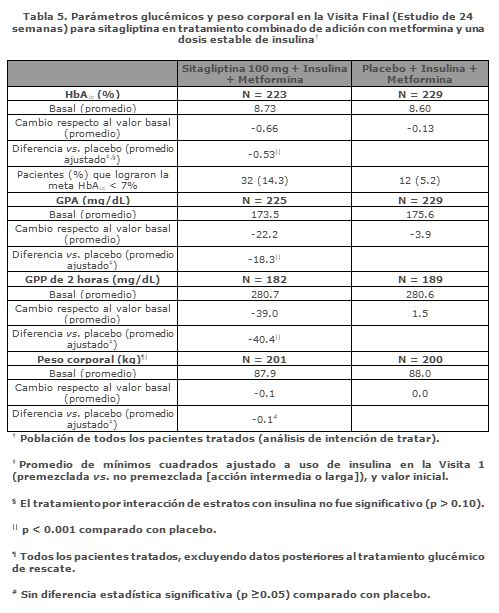
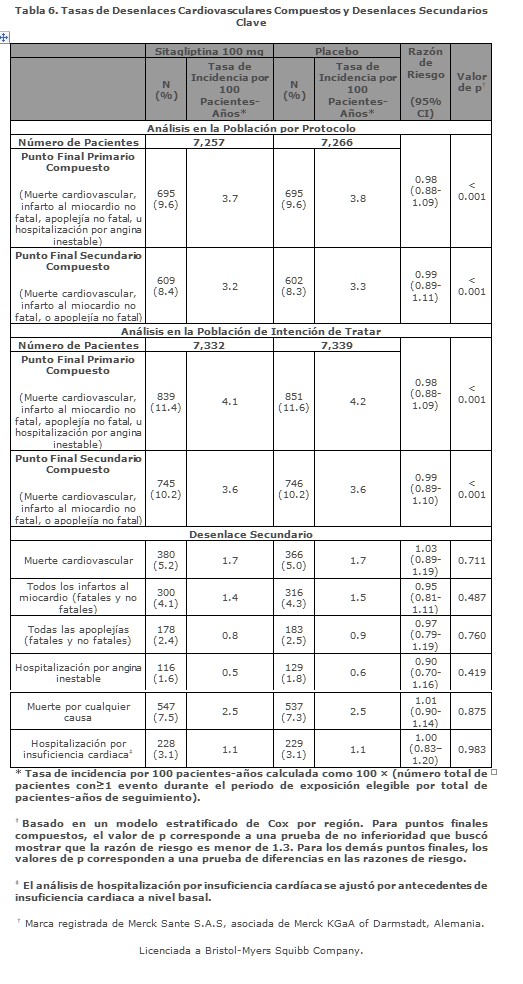
En otro estudio de 24-semanas, aleatorizado, doble-ciego, controlado con placebo, para evaluar la eficacia de la sitagliptina añadida a la insulina como terapia combinada, 660 pacientes con control glicémico inadecuado usando insulina glargina con o sin metformina (≥1,500 mg al día) fueron aleatorizados a la adición ya sea de 100 mg de sitagliptina (N = 330) o placebo (N = 330), administrados una vez al día mientras experimentaban intensificación de la terapia con insulina. Entre los pacientes que tomaban metformina, el valor inicial de HbA1c fue de 8.70% y la dosis inicial de insulina era 37 UI/día. Los pacientes recibieron instrucciones para calibrar su dosis de insulina glargina con base en los valores obtenidos de glucosa en ayuno con prueba por punción capilar. Los puntos finales glucémicos medidos incluyeron HbA1c y a FPG. Entre los pacientes que tomaban metformina, en la Semana 24, el aumento en la dosis diaria de insulina fue 21% menor en los pacientes tratados con sitagliptina (19UI/día, N = 285) que en los pacientes tratados con placebo (24 UI/día, N = 283). La diferencia en la dosis de insulina (-5 UI/día) fue estadísticamente significativa (p = 0.007). La reducción de HbA1c para pacientes tratados con sitagliptina, metformina, e insulina fue -1.35% comparado con -0.90% para pacientes tratados con placebo, metformina, e insulina, una diferencia de -0.45% [95% CI: -0.62, -0.29]. La reducción de FPG para pacientes tratados con sitagliptina, metformina, e insulina fue -54.8 mg/dL comparada con -43.0 mg/dL para pacientes tratados con placebo, metformina, e insulina, una diferencia de -11.8 mg/dL [95% CI: -18.7, -4.9]. La incidencia de hipoglucemia fue 24.9% para pacientes tratados con sitagliptina, metformina, e insulina y 37.8% para pacientes tratados con placebo, metformina e insulina. La diferencia en la incidencia de hipoglucemia (-12.9%) fue estadísticamente significativa (p < 0.001). Estudio controlado con activo (glipizida) en combinación con metformina: El efecto del tratamiento de mantenimiento a largo plazo se evaluó en un estudio de 52 semanas, doble ciego, controlado con glipizida en pacientes con diabetes tipo 2 inadecuadamente controlados con metformina sola ≥1,500 mg/día. En este estudio los pacientes fueron distribuidos al azar a la adición de sitagliptina 100 mg diarios (N = 588) o glipizida (N = 584) por 52 semanas. Los pacientes que recibieron glipizida iniciaron con una dosis de 5 mg al día y después se les ajustó la dosis a criterio del investigador hasta alcanzar una GPA objetivo de < 110 mg/dL, sin hipoglucemia significativa durante las siguientes 18 semanas. Como dosis máxima se permitió una dosificación de 20 mg al día para optimizar el control glucémico; después, la dosis de glipizida debía mantenerse constante. La dosis promedio de glipizida después del periodo de ajuste fue de 10.3 mg. Ambos tratamientos mejoraron significativamente el control de la glucemia respecto al inicio. Después de 52 semanas, la reducción en HbA1c respecto al valor basal fue -0.67% con sitagliptina 100 mg diarios y -0.67% con glipizida, lo cual confirmó una eficacia comparable de estos dos medicamentos. La reducción en GPA fue -10.0 mg/dL con sitagliptina y -7.5 mg/dL con glipizida. En un análisis post hoc, los pacientes con una HbA1c basal mayor (≥9%) tuvieron mayores reducciones en ambos grupos respecto al valor basal en HbA1c (sitagliptina, -1.68%; glipizida, -1.76%). En este estudio, la relación proinsulina a insulina, un marcador de eficiencia en la síntesis y liberación de insulina, mejoró con sitagliptina y se deterioró con el tratamiento con glipizida. La incidencia de hipoglucemia en el grupo tratado con sitagliptina (4.9%) fue significativamente menor que con el grupo con glipizida (32.0%). Los pacientes tratados con sitagliptina presentaron una disminución de peso promedio significativa, comparado con el aumento significativo de peso en los pacientes que recibieron glipizida (-1.5 kg vs. +1.1 kg). Clorhidrato de metformina: El estudio prospectivo con distribución al azar UKPDS (United Kingdom Prospective Diabetes Study) ha demostrado el beneficio a largo plazo del control intensivo de la glucemia en la diabetes tipo 2. El análisis de los resultados obtenidos en pacientes obesos tratados con metformina después de haber fracasado el tratamiento con dieta sola mostró:• una disminución significativa del riesgo absoluto de cualquier complicación relacionada con la diabetes en el grupo de metformina (29.8 casos/1,000 pacientes-años) en comparación con la dieta sola (43.3 casos/1,000 pacientes-años), p = 0.0023 y en comparación con los grupos de monoterapia con sulfonilurea y con insulina combinados (40.1 casos/1,000 pacientes-años) (p = 0.0034). • una disminución significativa del riesgo absoluto de mortalidad relacionada con la diabetes: Metformina, 7.5 muertes/1,000 pacientes-años; dieta sola, 12.7 muertes/1,000 pacientes-años (p = 0.017). • una disminución significativa del riesgo absoluto de mortalidad total: Metformina, 13.5 muertes/1,000 pacientes-años, en comparación con la dieta sola (20.6 muertes/1,000 pacientes-años) (p = 0.011), y en comparación con los grupos de monoterapia con sulfonilurea y con insulina combinados (18.9 muertes/1,000 pacientes-años) (p = 0.021). • una disminución significativa del riesgo absoluto de infarto del miocardio: Metformina, 11 casos/1,000 pacientes-años; dieta sola, 18 casos/1,000 pacientes-años (p = 0.01). Estudio de Seguridad Cardiovascular TECOS: El Estudio de Evaluación de Desenlaces Cardiovasculares (TECOS, por las siglas en inglés para Trial Evaluating Cardiovascular Outcomes with Sitagliptin) fue un estudio aleatorizado en 14,671 pacientes en la población de intención de tratar con una HbA1c de ≥ 6.5 a 8.0% con enfermedad CV establecida, que recibieron sitagliptina (7,332), 100 mg diarios (0.50 mg diarios si la TFG estimada basal era ≥30 y < 50 mL/min/1.73 m²) o placebo (7,339) añadidos al cuidado usual, apuntando a estándares regionales para HbA1c y factores de riesgo CV. No se incluyeron en el estudio pacientes con una TFG estimada < 30 mL/min/1.73 m². La población de estudio incluyó a 2,004 pacientes ≥75 años de edad y 3,324 pacientes con insuficiencia renal (TFG estimada < 60 mL/min/1.73 m²). Durante el curso del estudio, la diferencia promedio global estimada (SD) en HbA1c entre los grupos sitagliptina y placebo fue de 0.29% (0.01), 95% IC (-0.32, -0.27); p < 0.001. Los pacientes en el grupo sitagliptina recibieron menos agentes hipoglucemiantes que los del grupo placebo (razón de riesgo 0.72, 95% IC, 0.68 a
Contraindicaciones: VELMETIA (sitagliptina/metformina) está contraindicado en pacientes con:1. Insuficiencia renal severa (TFGe < 30 mL/min/1.73 m2) (Véase Precauciones generales, Clorhidrato de Metformina, Insuficiencia renal). 2. Hipersensibilidad conocida al fosfato de sitagliptina, clorhidrato de metformina o a cualquier otro componente de VELMETIA (véase Precauciones generales, Fosfato de sitagliptina, Reacciones de hipersensibilidad y Reacciones secundarias y adversas, Experiencia Después de la Comercialización). 3. Acidosis metabólica aguda o crónica, incluyendo cetoacidosis diabética con o sin coma. Se debe suspender temporalmente la administración de VELMETIA en los pacientes que van a ser sometidos a estudios radiológicos con administración intravascular de medios de contraste yodados, porque éstos pueden causar alteraciones agudas de la función renal (véase Precauciones generales, Clorhidrato de metformina). Restricciones de uso durante el embarazo y la lactancia: Embarazo: No hay estudios suficientes y bien controlados en mujeres embarazadas con VELMETIA o con sus componentes individuales; por lo tanto, no se conoce la seguridad de VELMETIA en ellas. Como otros agentes hipoglucemiantes orales, no se recomienda emplear VELMETIA durante el embarazo. No se han hecho estudios en animales con los componentes combinados de VELMETIA para evaluar sus efectos sobre la reproducción. Los datos siguientes están basados en los resultados de estudios realizados con sitagliptina o metformina solas. Fosfato de sitagliptina: La sitagliptina no fue teratogénica en ratas a dosis orales de hasta 250 mg/kg ni en conejas que recibieron hasta 125 mg/kg durante la organogénesis (dosis hasta 32 y 22 veces mayores, respectivamente, que la exposición en seres humanos, basándose en la dosis diaria recomendada en adultos de 100 mg diarios). En las ratas aumentó ligeramente la incidencia de malformaciones costales en los fetos (costillas ausentes, hipoplásicas u onduladas) con la dosificación de 1,000 mg/kg/día (aproximadamente 100 veces mayor que la exposición en seres humanos, basándose en la dosis diaria recomendada en adultos de 100 mg diarios). En las crías de las ratas que recibieron dosis orales de 1,000 mg/kg/día se observó una pequeña disminución del promedio de peso corporal antes del destete en ambos sexos y del promedio de aumento de peso después del destete en los machos. Sin embargo, los estudios de reproducción en animales no siempre pronostican la respuesta en los seres humanos. Clorhidrato de metformina: La metformina no fue teratogénica en ratas y conejas a dosificaciones de hasta 600 mg/kg/día, que representan una exposición aproximadamente dos y seis mayor, respectivamente, que la producida por la dosis diaria máxima recomendada en los seres humanos de 2,000 mg, basándose en el área de superficie corporal. Las concentraciones en los fetos demostraron una barrera placentaria parcial contra la metformina. Lactancia: No se han hecho estudios en hembras lactantes con los componentes combinados de VELMETIA. En estudios realizados con los componentes individuales, tanto la sitagliptina como la metformina fueron secretadas en la leche de las ratas lactantes. No se sabe si la sitagliptina es secretada con la leche humana. Por lo tanto, no se debe administrar VELMETIA a una mujer que está amamantando.
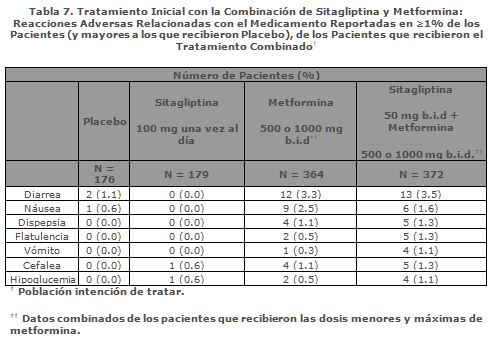
Reacciones secundarias y adversas: En estudios clínicos controlados con placebo en pacientes con diabetes mellitus tipo 2, la combinación de sitagliptina y metformina generalmente fue bien tolerada. La incidencia general de reacciones secundarias reportada por los pacientes que recibieron la combinación de sitagliptina y metformina fue similar a la reportada por los pacientes que recibieron la combinación de placebo y metformina. Tratamiento Combinado con Sitagliptina y Metformina: Tratamiento Inicial: En un estudio factorial de 24 semanas, controlado con placebo, de tratamiento inicial con sitagliptina 50 mg dos veces al día en combinación con metformina 500 ó 1,000 mg dos veces al día, los eventos adversos relacionados con el medicamento reportados por ≥1% (y mayor a los que recibieron placebo) de los pacientes que recibieron el tratamiento combinado se muestran en la Tabla 7.
Tratamiento Combinado de Adición a Metformina: En un estudio de 24 semanas, controlado con placebo, en el que se añadió sitagliptina a tratamiento en curso con metformina, 464 pacientes recibieron metformina y 100 mg una vez al día de sitagliptina y 237 recibieron placebo y metformina. La única reacción adversa reportada relacionada con el medicamento que ocurrió con una incidencia ≥1% y mayor que con placebo en los que recibieron sitagliptina y metformina fue náusea (1.1% con 100 mg de sitagliptina y metformina, 0.4% con placebo y metformina). Hipoglucemia y Reacciones Adversas Gastrointestinales: En los estudios controlados con placebo de tratamiento combinado con sitagliptina y metformina, la incidencia de hipoglucemia (independientemente de la causalidad atribuida por el investigador) reportada por los pacientes que recibieron la combinación de sitagliptina y metformina fue similar a la reportada por los pacientes tratados con metformina y placebo. La incidencia de reacciones adversas gastrointestinales preespecificadas en pacientes tratados con la combinación de sitagliptina y metformina fue similar a la reportada por los pacientes tratados con metformina sola; véase Tabla 8.
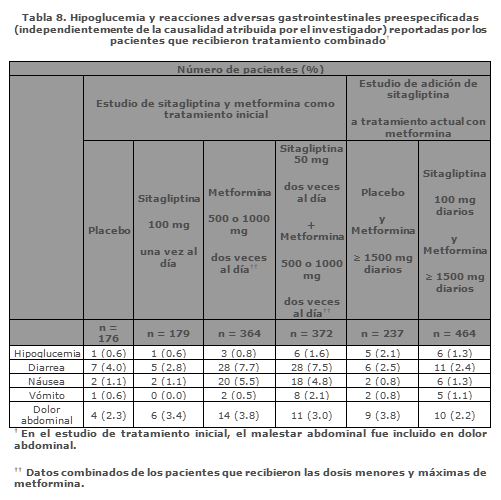
En todos los estudios las reacciones adversas de hipoglucemia se basaron en todos los reportes de hipoglucemia sintomática; no se requirió hacer una medición de la glucosa. Sitagliptina en Combinación con Metformina y una Sulfonilurea: En un estudio controlado con placebo de 24 semanas de sitagliptina 100 mg diarios añadidos a la combinación con glimepirida ≥ 4 mg diarios y metformina ≥1,500 mg diarios, las reacciones adversas relacionadas con el medicamento reportadas en ≥ 1% de los pacientes tratados con sitagliptina (n = 116) y más común que en los pacientes tratados con placebo (n = 113) fueron hipoglucemia (sitagliptina 13.8%, placebo 0.9%) y estreñimiento (1.7%, 0.0%). Sitagliptina en Combinación con Metformina y un Agonista PPARg: En un estudio controlado con placebo de sitagliptina 100 mg diarios añadidos al tratamiento combinado en curso de metformina y rosiglitazona, las reacciones adversas relacionadas con el medicamento reportadas durante el punto de tiempo primario a la semana 18 en ≥ 1% de los pacientes tratados con sitagliptina (n = 170) y más comúnmente que en los pacientes tratados con placebo (n = 92) fueron: cefalea (sitagliptina, 2.4%; placebo, 0.0%), diarrea (1.8%, 1.1%), náusea (1.2%, 1.1%), hipoglucemia (1.2%, 0.0%), y vómito (1.2%, 0.0%). A la semana 54, las reacciones adversas relacionadas con el medicamento reportadas en ≥ 1% de los pacientes tratados con sitagliptina y más comúnmente que en los pacientes tratados con placebo fueron: cefalea (2.4%, 0.0%), hipoglucemia (2.4%, 0.0%), infección del tracto respiratorio superior (1.8%, 0.0%), náusea (1.2%, 1.1%), tos (1.2%, 0.0%), infecciones fúngicas cutáneas (1.2%, 0.0%), edema periférico (1.2%, 0.0%), y vómito (1.2%, 0.0%). Sitagliptina en Combinación con Metformina e Insulina: En un estudio controlado con placebo de 24 semanas con sitagliptina 100 mg añadida en combinación al tratamiento en curso con metformina ≥ 1,500 mg diarios y una dosis estable de insulina, la única reacción adversa relacionada con el medicamento reportada en ≥1% de los pacientes tratados con sitagliptina (n = 229) y más comúnmente que en los pacientes tratados con placebo (n = 233) fue hipoglucemia (sitagliptina, 10.9%; placebo, 5.2%). En otro estudio de 24 semanas los pacientes recibieron sitagliptina como tratamiento adicional mientras se intensificaba la isulina (con o sin metformina), la única reacción adversa relacionada con el fármaco reportada en ≥1% de los pacientes tratados con sitagliptina y metformina y con mayor frecuencia que en los pacientes tratados con placebo y metformina fue el vómito (sitagliptina y metformina, 1.1%, placebo y metformina 0.4%). Pancreatitis: En un análisis combinado de 19 estudios clínicos doble ciego que incluyó datos de 10,246 pacientes distribuidos al azar a recibir sitagliptina 100 mg/día (N = 5,429) o el correspondiente control, tratamiento activo o placebo (N = 4,817), la incidencia de eventos no adjudicados de pancreatitis aguda fue 0.1 por 100 años-paciente en cada grupo (4 pacientes con un evento en 4,708 años-paciente para sitagliptina y 4 pacientes con un evento en 3,942 años-paciente para el grupo control). Ver también más adelante Estudio de Seguridad Cardiovascular TECOS (véase Precauciones generales, Pancreatitis). Con la combinación de sitagliptina y metformina no se observaron cambios clínicamente significativos en los signos vitales o en el electrocardiograma (incluyendo el intervalo QT). Reacciones Adversas Reportadas con Sitagliptina: No hubo reportes de reacciones adversas relacionadas con el medicamento con una incidencia ≥1% en los pacientes que recibieron sitagliptina. Reacciones Adversas Reportadas con Metformina: Las reacciones adversas reportadas (independientemente de la causalidad) en más del 5% de los pacientes tratados con metformina y con más frecuencia que en los pacientes tratados con placebo son diarrea, náusea/vómito, flatulencia, astenia, indigestión, malestar abdominal y cefalea. Estudio de Seguridad Cardiovascular TECOS: El Estudio para Evaluar los Desenlaces Cardiovasculares con Sitagliptina (The Trial Evaluating Cardiovascular Outcomes with Sitagliptin, TECOS) incluyó a 7,332 pacientes tratados con sitagliptina, a 100 mg diarios (o 50 mg diarios si la tasa de filtración glomerular basal estimada (TFGe) era ≥30 y < 50 mL/min/1.73 m²), y 7,339 pacientes tratados con placebo en la población de intención de tratar. Ambos tratamientos se añadieron al cuidado usual, apuntando a estándares regionales para HbA1c y factores de riesgo CV. La población de estudio incluyó un total de 2,004 pacientes ≥75 años de edad (970 tratados con sitagliptina y 1,034 tratados con placebo). La incidencia global de eventos adversos graves en los pacientes que recibieron sitagliptina fue similar a la de los pacientes que recibieron placebo. La evaluación de complicaciones relacionadas con la diabetes pre-especificadas reveló incidencias similares entre los grupos, incluyendo infecciones (18.4% de los pacientes tratados con sitagliptina y 17.7% de los pacientes tratados con placebo) e insuficiencia renal (1.4% de los pacientes tratados con sitagliptina y 1.5% de los pacientes tratados con placebo). El perfil de eventos adversos en los pacientes ≥75 años de edad fue generalmente similar al de la población general. En la población de intención de tratar, entre los pacientes que estaban usando insulina y/o una sulfonilurea a nivel basal, la incidencia de hipoglucemia grave fue de 2.7% en los pacientes tratados con sitagliptina y 2.5% en los pacientes tratados con placebo, entre los pacientes que no estaban usando insulina y/o una sulfonilurea a nivel basal, la incidencia de hipoglucemia grave fue de 1.0% en los pacientes tratados con sitagliptina y de 0.7% en los pacientes tratados con placebo. La incidencia de eventos de pancreatitis con adjudicación confirmada fue de 0.3% en los pacientes tratados con sitagliptina y de 0.2% en los pacientes tratados con placebo. La incidencia de eventos de malignidad con adjudicación confirmada fue de 3.7% en los pacientes tratados con sitagliptina y de 4.0% en los pacientes tratados con placebo. Experiencia Después de la Comercialización: Durante el uso después de la comercialización de VELMETIA, o sitagliptina, uno de los componentes de VELMETIA, se han identificado reacciones adversas adicionales. Estas reacciones han sido reportadas cuando VELMETIA o sitagliptina se utilizaron como monoterapia o en combinación con otros medicamentos hipoglucemiantes. Como estas reacciones se reportaron voluntariamente de una población de tamaño incierto, generalmente no es posible estimar de manera confiable su frecuencia o establecer una relación causal a la exposición del medicamento. Reacciones de hipersensibilidad incluyendo anafilaxis, angioedema, erupción cutánea, urticaria, vasculitis cutánea, y condiciones cutáneas exfoliativas incluyendo el síndrome de Stevens-Johnson (véase Contraindicaciones y Precauciones generales, Fosfato de sitagliptina, Reacciones de hipersensibilidad); pancreatitis aguda, incluyendo pancreatitis necrotizante o hemorrágica fatal y no fatal (véase Precauciones generales, Pancreatitis); deterioro de la función renal, incluyendo insuficiencia renal aguda (algunas veces requiriendo diálisis); penfigoide ampolloso (véase Precauciones generales, penfigoide ampolloso) infección de vías respiratorias superiores; nasofaringitis; estreñimiento, vómito, cefalea; artralgia, mialgia, dolor en las extremidades, dolor de espalda, prurito.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: Fosfato de sitagliptina: Se hizo un estudio de dos años sobre la carcinogenicidad en ratas machos y hembras que recibieron dosis orales de sitagliptina de 50, 150 y 500 mg/kg/día. Con la dosificación mayor (exposición aproximadamente 58 veces mayor que en los seres humanos, basándose en la dosis diaria recomendada en adultos de 100 mg diarios) hubo un aumento de la incidencia de adenomas y carcinomas hepáticos en los machos y de carcinomas hepáticos en las hembras. Esa dosificación se asoció con hepatotoxicidad en las ratas. La dosificación con la que no se observó ninguna inducción de neoplasias hepáticas fue la de 150 mg/kg/día (aproximadamente 19 veces mayor que la exposición en seres humanos con la dosis recomendada de 100 mg diarios). Como se ha mostrado que en las ratas la hepatotoxicidad está correlacionada con la inducción de neoplasias hepáticas, ese aumento de la incidencia de tumores hepáticos es probablemente secundario a toxicidad hepática crónica a esa alta dosificación. Se desconoce la importancia clínica de esos resultados para los seres humanos. Se hizo un estudio de dos años sobre la carcinogenicidad en ratones machos y hembras, con dosis orales de sitagliptina de 50, 125, 250 y 500 mg/kg/día. La sitagliptina no aumentó la incidencia de tumores de ningún órgano en los ratones con dosis de hasta 500 mg/kg/día (exposición aproximadamente 68 veces mayor que la usada en seres humanos por la dosis recomendada de 100 mg diarios). Clorhidrato de metformina: Se han hecho estudios a largo plazo sobre la carcinogenicidad en ratas (durante 104 semanas) y en ratones (durante 91 semanas) con dosificaciones de hasta 900 mg/kg/día y 1,500 mg/kg/día respectivamente, que son aproximadamente cuatro veces mayores que la dosis diaria máxima de 2,000 mg recomendada en seres humanos basándose en la comparación de las áreas de superficie corporal. No se encontró ningún indicio de carcinogenicidad de la metformina en los ratones machos o hembras ni en las ratas machos, pero sí aumentó la incidencia de pólipos benignos del estroma uterino en las ratas hembras tratadas con 900 mg/kg/día. Mutagénesis: Fosfato de sitagliptina: La sitagliptina no fue mutagénica ni clastogénica en una serie de estudios de toxicología genética que incluyeron la prueba bacteriana de Ames (prueba de mutagénesis microbiana), la prueba de aberraciones cromosómicas en células de ovario de hámster Chino (células CHO) y una prueba citogenética in vitro con células CHO, un ensayo de dilución alcalina en ADN de hepatocitos de rata (que mide la capacidad del medicamento para inducir roturas de una cadena en el ADN), y una prueba de micronúcleos in vivo. Clorhidrato de metformina: No hubo ningún indicio de potencial mutagénico de la metformina en las siguientes pruebas in vitro: Prueba de Ames (S. typhimurium), prueba de mutación genética (en células de linfoma de ratón) o prueba de aberraciones cromosómicas (en linfocitos humanos). Los resultados de la prueba de micronúcleos in vivo en ratones también fueron negativos. Reproducción: Fosfato de sitagliptina: No se observó ningún efecto adverso sobre la fertilidad en ratas machos y hembras que recibieron dosis orales de hasta 1,000 mg/kg de sitagliptina (exposición hasta unas 100 veces mayor que la exposición en seres humanos, basándose en la dosis diaria recomendada en adultos de 100 mg diarios) antes y durante el apareamiento. Clorhidrato de metformina: La fertilidad de las ratas machos o hembras no fue afectada por la metformina administrada a dosificaciones tan altas como 600 mg/kg/día, que es aproximadamente tres veces mayor que la dosis diaria máxima recomendada en seres humanos basándose en la comparación de las áreas de superficie corporal. Desarrollo fetal: Fosfato de sitagliptina: La sitagliptina no fue teratogénica en ratas a dosis orales de hasta 250 mg/kg ni en conejas que recibieron hasta 125 mg/kg durante la organogénesis (exposición hasta 32 y 22 veces mayor, respectivamente, que la causada en seres humanos por la dosis diaria recomendada en adultos de 100 mg diarios). En las ratas aumentó ligeramente la incidencia de malformaciones costales en los fetos (costillas ausentes, hipoplásicas u onduladas) con la dosificación de 1,000 mg/kg/día (exposición aproximadamente 100 veces mayor que la causada en seres humanos por la dosis diaria recomendada en adultos de 100 mg diarios). El nivel de dosificación con el que no se observaron efectos sobre el desarrollo fetal fue 250 mg/kg/día (exposición 32 veces mayor que la causada en seres humanos por la dosis diaria recomendada en adultos de 100 mg diarios). En las crías de las ratas que recibieron dosis orales de 1,000 mg/kg/día se observaron disminuciones relacionadas con el tratamiento del promedio de peso corporal antes del destete en ambos sexos y del promedio de aumento de peso después del destete en los machos.
Interacciones medicamentosas y de otro género: Sitagliptina y metformina: La coadministración de dosis múltiples de 50 mg de sitagliptina dos veces al día y 1,000 mg de metformina dos veces al día no alteró significativamente la farmacocinética de ninguno de los dos medicamentos en pacientes con diabetes tipo 2. No se han hecho estudios de interacción farmacocinética con VELMETIA, pero sí con los componentes individuales de VELMETIA (fosfato de la sitagliptina y clorhidrato de metformina. Fosfato de sitagliptina: En los estudios de interacciones farmacológicas, la sitagliptina no tuvo efectos de importancia clínica sobre la farmacocinética de los siguientes medicamentos: Metformina, rosiglitazona, gliburida, simvastatina, warfarina y anticonceptivos orales. Según esos datos, la sitagliptina no inhibe las isoenzimas CYP3A4, 2C8 y 2C9 del citocromo P-450. Y basándose en los datos in vitro, no es de esperarse que inhiba las isoenzimas CYP2D6, 1A2, 2C19 y 2B6, ni que induzca a CYP3A4. Se han realizado análisis de farmacocinética de población en pacientes con diabetes tipo 2. Los medicamentos administrados concomitantemente no tuvieron un efecto clínicamente importante en la farmacocinética de sitagliptina. Los medicamentos evaluados fueron aquellos que comúnmente se administran en pacientes con diabetes tipo 2, incluyendo reductores de colesterol (p. ej., estatinas, fibratos, ezetimiba), antiplaquetarios (clopidogrel), antihipertensivos (inhibidores de la ECA, bloqueadores de los receptores de angiotensina, bloqueadores beta, antagonistas de canales de calcio, hidroclorotiazida), analgésicos y anti-inflamatorios no esteroideos (p. ej., naproxeno, diclofenaco, celecoxib), antidepresivos (p. ej., bupropión, fluoxetina, sertralina), antihistamínicos (cetrizina), inhibidores de la bomba de protones (p. ej., omeprazol, lansoprazol) y medicamentos para disfunción eréctil (p. ej., sildenafil). Al coadministrar sitagliptina y digoxina, aumentaron ligeramente el área bajo la curva de concentración (ABC, 11%) y el promedio de concentración máxima (Cmáx, 18%) de la digoxina. No se considera que esos aumentos tengan importancia clínica. Se debe vigilar apropiadamente a los pacientes que estén recibiendo digoxina. El ABC y la Cmáx de la sitagliptina aumentaron aproximadamente 29% y 68%, respectivamente, al coadministrar dosis orales únicas de 100 mg de sitagliptina* y de 600 mg de ciclosporina, que es un potente inhibidor de prueba de la p-glucoproteína. No se considera que esos cambios observados en la farmacocinética de la sitagliptina tengan importancia clínica. Clorhidrato de metformina: Gliburida: En un estudio sobre interacción de dosis únicas en pacientes con diabetes tipo 2, la coadministración de metformina y gliburida no causó ningún cambio en la farmacocinética o la farmacodinámica de la metformina. Se observaron disminuciones del ABC y de la Cmáx de la gliburida, pero esas disminuciones fueron muy variables. Debido a que fue un estudio con dosis únicas y a la falta de correlación entre las concentraciones sanguíneas de gliburida y los efectos farmacodinámicos, es incierta la importancia clínica de esta interacción. Furosemida: Un estudio sobre la interacción de dosis únicas de metformina y furosemida en sujetos sanos demostró que la coadministración afectó los parámetros farmacocinéticos de ambos compuestos. La furosemida aumentó 22% la Cmáx plasmática y sanguínea y 15% el ABC sanguínea de la metformina, sin ningún cambio significativo en su depuración renal. Cuando se coadministró con metformina, la Cmáx y el ABC de la furosemida fueron 31% y 12% menores, respectivamente, que cuando se administró sola, y su vida media terminal disminuyó 32%, sin ningún cambio significativo en su depuración renal. No hay información disponible acerca de la interacción de la metformina y la furosemida cuando se coadministran por tiempo prolongado. Nifedipina: Un estudio sobre la interacción de dosis únicas de metformina y nifedipina en sujetos sanos demostró que la coadministración de nifedipina aumentó 20% la Cmáx y 9% el ABC de la metformina, así como la cantidad de metformina excretada en la orina. Su Tmáx y su vida media no se modificaron. La nifedipina aumenta la absorción de metformina, y ésta tuvo efectos mínimos sobre la nifedipina. El uso concomitante de medicamentos que interfieren con los sistemas comunes de transporte tubular renal, involucrados en la eliminación renal de la metformina (p. ej. Transportador-2 de cationes orgánicos [OCT2]/inhibidores de extrusión de múltiples fármacos y toxinas [MATE] tales como ranolazina, vandetanib, dolutegravir y cimetidina) pueden incrementar la exposición sistémica a la metformina y puede aumentar el riesgo de acidosis láctica. Se debe considerar los beneficios y los riesgos de tratamientos concomitantes. Otros: Ciertos medicamentos tienden a producir hiperglucemia y pueden impedir el control de la glucemia. Esos medicamentos incluyen las tiazidas y otros diuréticos, corticosteroides, fenotiacinas, productos tiroideos, estrógenos, anticonceptivos orales, fenitoína, ácido nicotínico, simpaticomiméticos, bloqueadores de canales de calcio y la isoniacida. Cuando se administra alguno de esos medicamentos a un paciente que está tomando VELMETIA, se le debe vigilar estrechamente para controlar bien su glucemia. En estudios de interacción con dosis únicas en voluntarios sanos, la coadministración de metformina y propranolol o de metformina e ibuprofeno no afectó la farmacocinética de esos medicamentos. A diferencia de las sulfonilureas, que se unen en gran proporción a las proteínas plasmáticas, la metformina se une escasamente a ellas, por lo que es menos probable que interactúe con medicamentos que se unen mucho a las proteínas plasmáticas, como los salicilatos, las sulfonamidas, el cloranfenicol y el probenecid.
Alteraciones en los resultados de pruebas de laboratorio: Fosfato de sitagliptina: La incidencia de reacciones adversas de laboratorio fue similar en los pacientes tratados con sitagliptina y metformina comparado con los pacientes tratados con placebo y metformina. A lo largo de esos estudios se observó un pequeño aumento del número de leucocitos (una diferencia de aproximadamente 200 leucocitos/ml en comparación con el placebo; cuenta promedio basal de leucocitos de aproximadamente 6,600/ml), debido a un pequeño aumento de los neutrófilos. Esta observación se ha hecho en la mayoría pero no en todos los estudios. Este cambio en los parámetros de laboratorio no se considera clínicamente importante. Clorhidrato de metformina: En estudios clínicos comparativos de metformina de 29 semanas de duración se observó una disminución a valores subnormales de las concentraciones séricas previas normales de vitamina B12, sin manifestaciones clínicas, en aproximadamente 7% de los pacientes. Sin embargo, esa disminución (debida posiblemente a la interferencia de la absorción de esa vitamina a partir del complejo B12-factor intrínseco) muy rara vez se acompaña de anemia y parece ser rápidamente reversible al suspender la administración de metformina o al administrar vitamina B12 (véase Precauciones generales, Clorhidrato de metformina).
Precauciones generales: No se debe usar VELMETIA® en pacientes con diabetes tipo 1 ni para tratar la cetoacidosis diabética. Pancreatitis: En pacientes que toman sitagliptina ha habido reportes de pancreatitis aguda, incluyendo pancreatitis necrotizante o hemorrágica fatal y no fatal (véase Reacciones secundarias y adversas, Experiencia después de la comercialización). Se debe advertir a los pacientes sobre los síntomas característicos de la pancreatitis aguda: dolor abdominal persistente y severo. Se ha observado que la pancreatitis se resuelve tras la suspensión de sitagliptina. Si se sospecha pancreatitis, se debe descontinuar VELMETIA o cualquier otro medicamento que se sospeche pueda causarla. Vigilancia de la función renal: Se sabe que la metformina y la sitagliptina son excretadas en gran parte por vía renal. El riesgo de acumulación de metformina y acidosis láctica aumenta con el grado de insuficiencia renal. VELMETIA está contraindicado en insuficiencia renal severa, pacientes con una TFGe < 30 mL/min/1.73 m² (Véase Dosis y vía de administración, Contraindicaciones y Precauciones generales, Clorhidrato de Metformina, Acidosis Láctica). Antes de iniciar el tratamiento con VELMETIA, y después por lo menos cada año, se debe evaluar la función renal. Si se prevé el desarrollo de insuficiencia renal, se debe medir la función renal con mayor frecuencia y suspender la administración de VELMETIA® si aparecen signos de insuficiencia renal. Hipoglucemia en combinación con una sulfonilurea o con insulina: Como es típico con otros medicamentos hipoglucemiantes, se ha observado hipoglucemia en pacientes que son tratados con sitagliptina y metformina en combinación con insulina o una sulfonilurea (véase Reacciones secundarias y adversas). Por ello, para reducir el riesgo de hipoglucemia inducida por sulfonilurea o por insulina, se debe considerar una dosis menor de la sulfonilurea o de la insulina (véase Dosis y vía de administración). Fosfato de sitagliptina: Hipoglucemia en combinación con una sulfonilurea o con insulina: En los estudios clínicos de sitagliptina como monoterapia o como parte del tratamiento combinado con medicamentos que se sabe causan hipoglucemia (p. ej. metformina o un agonista del PPARg [tiazolidinediona]), las tasas de hipoglucemia fueron similares a las observadas en los pacientes que recibieron placebo. Como es típico con otros medicamentos hipoglucemiantes, se ha observado hipoglucemia en pacientes que son tratados con sitagliptina en combinación con insulina o una sulfonilurea (véase Reacciones secundarias y adversas). Por ello, para reducir el riesgo de hipoglucemia inducida por sulfonilurea o por insulina, se debe considerar una dosis menor de la sulfonilurea o de la insulina (véase Dosis y vía de administración). Reacciones de hipersensibilidad: Ha habido reportes después de la comercialización de reacciones de hipersensibilidad graves en pacientes tratados con sitagliptina, uno de los componentes de VELMETIA. Estas reacciones incluyen anafilaxis, angioedema, y condiciones cutáneas exfoliativas, incluyendo el síndrome de Stevens-Johnson. Como estas reacciones se reportan voluntariamente de una población de tamaño incierto, en general no es posible estimar de manera confiable su frecuencia o establecer una relación causal a la exposición al fármaco. El inicio de estas reacciones ocurrió dentro de los primeros 3 meses después del inicio del tratamiento con sitagliptina, algunos reportes ocurrieron después de la primera dosis. Si se sospecha una reacción de hipersensibilidad, descontinúe VELMETIA, evalúe otras causas potenciales del evento e inicie un tratamiento alternativo para la diabetes (véase Contraindicaciones y Reacciones secundarias y adversas, Experiencia Después de la Comercialización). Penfigoide ampolloso: En contexto de post-comercialización se han reportado casos de penfigoide ampolloso que requirieron hospitalización con el uso de inhibidores de la DPP-4. En estos casos reportados, los pacientes típicamente se recuperaron con tratamiento inmunosupresor tópico o sistémico y suspendiendo el tratamiento con el inhibidor de la DPP-4. Pida a sus pacientes reportar la aparición de ampollas o erosiones cuando estén bajo tratamiento con VELMETIA. En caso de sospecha de penfigoide ampolloso se debe suspender el tratamiento con VELMETIA y se debe considerar referir al paciente con el dermatólogo para diagnóstico y tratamiento apropiado. Clorhidrato de metformina:Acidosis láctica: La acidosis láctica es una complicación rara, pero grave, que puede ocurrir debido a la acumulación de metformina durante el tratamiento con VELMETIA (fosfato de sitagliptina/clorhidrato de metformina); es mortal en aproximadamente 50% de los casos. También puede ocurrir en varios estados fisiopatológicos, incluyendo diabetes mellitus, y cuando hay hipoperfusión e hipoxemia tisulares importantes. La acidosis láctica se caracteriza por concentraciones sanguíneas elevadas de lactato ( > 5 mmol/L), disminución del pH sanguíneo, trastornos electrolíticos con aumento de la brecha aniónica, y aumento de la relación lactato/piruvato. Cuando la metformina está implicada como causa de la acidosis láctica, generalmente sus concentraciones plasmáticas son mayores de 5 mg/mL. La incidencia reportada de acidosis láctica en pacientes que están recibiendo clorhidrato de metformina es muy baja (aproximadamente 0.03 casos/1,000 pacientes-años y aproximadamente 0.015 casos mortales por cada 1,000 pacientes/años). En más de 20,000 pacientes-años de exposición a la metformina en los estudios clínicos, no hubo ningún reporte de acidosis láctica. Los casos reportados han ocurrido principalmente en pacientes diabéticos con insuficiencia renal significativa, incluyendo tanto nefropatía intrínseca como hipoperfusión renal, a menudo en un cuadro de múltiples problemas médicos/quirúrgicos concomitantes y numerosos medicamentos (Véase Dosis y vía de administración, Recomendaciones de uso para pacientes con Insuficiencia Renal). Los pacientes con insuficiencia cardiaca congestiva que necesitan tratamiento farmacológico, en particular aquellos con insuficiencia cardiaca congestiva inestable o aguda que están en riesgo de hipoperfusión e hipoxemia, tienen mayor riesgo de presentar acidosis láctica. El riesgo de acidosis láctica aumenta con el grado de insuficiencia renal y con la edad del paciente; por lo tanto, ese riesgo podría disminuir significativamente vigilando regularmente la función renal de los pacientes que reciben metformina y usando la dosis mínima efectiva de ésta, particularmente en los pacientes de edad avanzada (Véase más abajo Empleo en Pacientes de Edad Avanzada). Además, se debe suspender enseguida la administración de metformina si aparece cualquier trastorno asociado con hipoxemia, deshidratación o sepsis. Debido a que el deterioro de la función hepática puede limitar significativamente la capacidad para depurar lactato, generalmente se debe evitar administrar metformina a pacientes con signos clínicos o de laboratorio de hepatopatía. Los pacientes que están tomando metformina deben evitar el consumo excesivo agudo o crónico de alcohol, porque éste potencia los efectos de la metformina sobre el metabolismo del lactato. Además, se debe suspender temporalmente la administración de metformina antes de cualquier estudio radiológico con un medio de contraste intravascular o de cualquier intervención quirúrgica. En muchos casos la acidosis láctica inicia sutilmente, acompañada sólo por síntomas inespecíficos como malestar general, mialgias, disnea, incremento de la somnolencia y malestar abdominal inespecífico. Con una acidosis mayor se pueden asociar hipotermia, hipotensión y bradiarritmias resistentes. El paciente y su médico deben tener presente la posible importancia de esos síntomas, se debe indicar al paciente que informe inmediatamente a su médico si aparecen, y se debe suspender la administración de metformina hasta aclarar la situación. Puede ser útil medir los electrólitos, cetonas séricos, glucemia y, si está indicado, el pH sanguíneo, el lactato y la concentración sanguínea de metformina. Una vez que se ha estabilizado a un paciente con determinada dosificación de metformina, es improbable que los síntomas gastrointestinales, que son comunes durante el inicio del tratamiento, estén relacionados con el medicamento. La aparición posterior de síntomas gastrointestinales puede ser debida a acidosis láctica o a otra enfermedad grave.Las concentraciones de lactato en plasma venoso en ayunas mayores que el límite superior normal, pero menores de 5 mmol/L en pacientes que están tomando metformina no indican necesariamente una inminente acidosis láctica, sino que pueden ser debidas a otros mecanismos, como diabetes mal controlada, obesidad, actividad física intensa o problemas técnicos en el manejo de las muestras. Se debe sospechar acidosis láctica en cualquier paciente diabético con acidosis metabólica, pero sin signos de cetoacidosis (cetonuria y cetonemia). La acidosis láctica es una urgencia médica que se tiene que tratar en un hospital. En el caso de un paciente con acidosis láctica que está tomando metformina, se debe suspender inmediatamente la administración de ésta e iniciar enseguida medidas de sostén. Dado que el clorhidrato de metformina es dializable (con una depuración de hasta 170 mL/min con buenas condiciones hemodinámicas), se recomienda una pronta hemodiálisis para corregir la acidosis y extraer la metformina acumulada. En muchos casos ese tratamiento corrige rápidamente los síntomas y acelera la recuperación (véase Contraindicaciones).Hipoglucemia: En las circunstancias usuales de uso, los pacientes que reciben metformina sola no presentan hipoglucemia, pero ésta puede ocurrir cuando el ingreso calórico es deficiente, cuando la actividad física intensa no es compensada por complementos calóricos, o durante el uso concomitante de otros agentes que disminuyen la glucemia (como las sulfonilureas y la insulina) o de etanol. Los pacientes ancianos, debilitados, desnutridos, con insuficiencia suprarrenal o hipofisiaria o con intoxicación alcohólica son particularmente propensos a los efectos hipoglucemiantes. La hipoglucemia puede ser difícil de identificar en los ancianos y en las personas que están tomando bloqueadores ß-adrenérgicos. Uso concomitante de medicamentos que pueden afectar la función renal o la disposición de la metformina: Se deben usar con precaución los medicamentos concomitantes que pueden afectar la función renal, causar cambios hemodinámicos significativos o interferir con la disposición de la metformina, como medicamentos catiónicos que son eliminados por secreción tubular (véase Interacciones medicamentosas, Clorhidrato de metformina). Estudios radiológicos con medios de contraste yodados intravasculares, como urografía o colangiografía intravenosas, angiografía, y tomografía computarizada con medio de contraste intravascular: Los medios de contraste intravasculares yodados pueden causar alteraciones agudas de la función renal, y se han asociado con acidosis láctica en pacientes que recibían metformina (véase Contraindicaciones). Por lo tanto, en pacientes con una TFGe de ≥30 a < 60 mL/min/1.73 m², en pacientes con antecedente de insuficiencia hepática, alcoholismo o insuficiencia cardiaca, o en pacientes que recibirán contraste yodado intra-arterial, se debe suspender temporalmente la administración de VELMETIA antes o al momento y durante 48 horas después de hacer cualquiera de esos estudios, y continuarla sólo después de reevaluar la función renal y comprobar que es aceptable (véase Dosis y vía de administración). Estados hipóxicos: El colapso cardiovascular (choque) de cualquier causa, la insuficiencia cardiaca congestiva aguda, el infarto agudo del miocardio y otros trastornos caracterizados por hipoxemia se han asociado con acidosis láctica y también pueden causar hiperazoemia prerrenal. Si ocurren en pacientes bajo tratamiento con VELMETIA, se debe suspender enseguida la administración de este medicamento. Intervenciones quirúrgicas: Se debe suspender temporalmente la administración de VELMETIA antes de cualquier intervención quirúrgica (excepto en operaciones menores en las que no se restringe la ingestión de alimentos y líquidos), y no se debe reiniciar hasta que el paciente haya vuelto a alimentarse por vía oral y se haya comprobado que su función renal es normal. Ingestión de alcohol: Se sabe que el alcohol potencia el efecto de la metformina sobre el metabolismo del lactato, por lo que se debe advertir a los pacientes que deben evitar el consumo excesivo agudo o crónico de alcohol mientras están recibiendo VELMETIA. Deterioro de la función hepática: Como el deterioro de la función hepática se ha asociado con algunos casos de acidosis láctica, generalmente se debe evitar administrar VELMETIA a pacientes con signos clínicos o de laboratorio de hepatopatía. Concentraciones de v
Dosis y vía de administración: Vía de administración: Oral. General: La dosificación del tratamiento ahipoglucemiante con VELMETIA se debe individualizar basándose en el tratamiento actual del paciente, la eficacia y la tolerabilidad, sin exceder las dosis diarias máximas recomendadas de 100 mg de sitagliptina. Generalmente VELMETIA se debe administrar dos veces al día con alimentos, aumentando gradualmente la dosis para disminuir los efectos colaterales gastrointestinales asociados a metformina. Recomendaciones sobre la dosificación: La dosis inicial de VELMETIA se debe basar en el tratamiento actual del paciente. VELMETIA se debe administrar dos veces al día con alimentos. Los comprimidos de VELMETIA están disponibles en:• 50 mg de sitagliptina/500 mg de clorhidrato de metformina.• 50 mg de sitagliptina/850 mg de clorhidrato de metformina. • 50 mg de sitagliptina/1000 mg de clorhidrato de metformina. Tratamiento inicial: En pacientes con diabetes tipo 2 con hiperglucemia inadecuadamente controlada con dieta y ejercicio solos, la dosis inicial total diaria recomendada de VELMETIA es de 100 mg de sitagliptina/1000 mg de clorhidrato de metformina. En los pacientes con control glucémico inadecuado a esta dosis, se puede escalar gradualmente para reducir los efectos adversos asociados con metformina, hasta la dosis máxima diaria recomendada de metformina de 2,000 mg. Para pacientes inadecuadamente controlados con la monoterapia con metformina: En los pacientes en los que metformina sola no controla suficientemente la glucemia, la dosis inicial total recomendada de VELMETIA es de 100 mg de sitagliptina y la dosis previamente prescrita de metformina. Para pacientes inadecuadamente controlados con la monoterapia con sitagliptina: En los pacientes en los que sitagliptina sola no controla suficientemente la glucemia, la dosificación inicial recomendada de VELMETIA es de 100 mg de sitagliptina/1000 mg de clorhidrato de metformina. La dosis de metformina puede ser escalada gradualmente conforme sea necesario para lograr el control glucémico. Se debe considerar escalar gradualmente la dosis para reducir los efectos adversos gastrointestinales asociados con metformina. A los pacientes que están tomando sitagliptina sola a una dosificación ajustada por insuficiencia renal no se les debe cambiar a VELMETIA (véase Contraindicaciones). Para pacientes que están tomando sitagliptina coadministrada con metformina: En los pacientes que están tomando sitagliptina y metformina por separado, se puede iniciar la administración de VELMETIA a la dosis previamente prescrita de sitagliptina y metformina. Para pacientes inadecuadamente controlados con la combinación de tratamiento dual con cualquiera de dos de los siguientes tres medicamentos; sitagliptina, metformina o una sulfonilurea: La dosificación inicial usual de VELMETIA debe procurar 50 mg de sitagliptina dos veces al día (100 mg dosis total diaria de sitagliptina). Para determinar la dosis inicial del componente metformina se debe considerar el nivel de control glucémico del paciente y la dosis actual (si la hay) de metformina. Para reducir los efectos gastrointestinales asociados a metformina se debe considerar ajustar la dosis gradualmente. Los pacientes que inician o que actualmente toman una sulfonilurea pueden requerir una dosis menor de la sulfonilurea para reducir el riesgo de hipoglucemia inducida por sulfonilureas (véase Precauciones generales). Para pacientes inadecuadamente controlados con la combinación de tratamiento dual con cualquiera de dos de los siguientes tres medicamentos: sitagliptina, metformina o un agonista del PPARc (por ejemplo, tiazolidinedionas): La dosificación inicial usual de VELMETIA debe procurar 100 mg dosis total diaria de sitagliptina. Para determinar la dosis inicial del componente metformina se debe considerar el nivel de control glucémico del paciente y la dosis actual (si la hay) de metformina. Para reducir los efectos gastrointestinales asociados a metformina se debe considerar ajustar la dosis gradualmente. Para pacientes inadecuadamente controlados con la combinación de tratamiento dual con cualquiera de dos de los siguientes tres medicamentos: sitagliptina, metformina o insulina: La dosificación inicial usual de VELMETIA debe procurar 100 mg dosis total diaria de sitagliptina. Para determinar la dosis inicial del componente metformina se debe considerar el nivel de control glucémico del paciente y la dosis actual (si la hay) de metformina. Para reducir los efectos gastrointestinales asociados a metformina se debe considerar ajustar la dosis gradualmente. Los pacientes con insulina o que la están iniciando pueden requerir dosis menores de ésta para reducir el riesgo de hipoglucemia (véase Precauciones generales). No se han hecho estudios examinando específicamente la seguridad y la eficacia de VELMETIA en pacientes que estaban siendo tratados con otros agentes hipoglucemiantes y fueron cambiados a VELMETIA. Cualquier cambio en el tratamiento de la diabetes tipo 2 se debe hacer con precaución y vigilancia adecuada, porque pueden ocurrir cambios en el control de la glucemia. Recomendaciones de uso en pacientes con Insuficiencia Renal: Se debe evaluar la función renal antes de iniciar el tratamiento con VELMETIA y periódicamente después de iniciarlo. VELMETIA está contraindicado en pacientes con una TFGe < 30 mL/min/1.73 m². El uso de VELMETIA no se recomienda en pacientes con una TFGe ≥ 30 mL/min/1.73 m² y < 45 mL/min/1.73 m² debido a que estos pacientes requieren una dosis inferior de sitagliptina, la cual, no está disponible para la presentación combinada de VELMETIA. Interrupción del tratamiento con VELMETIA por procedimientos de imagenología con contraste yodado: Se debe descontinuar VELMETIA al momento, o previo a un procedimiento de imagenología que involucre contraste yodado en pacientes con una TFGe de ≥ 30 a < 60 mL/min/1.73 m², en pacientes con antecedente de enfermedad hepática, alcoholismo o insuficiencia cardiaca, o en pacientes que recibirán contraste yodado intra-arterial. Se debe reevaluar la TFGe a las 48 horas posterior al procedimiento de imagenología, el tratamiento con VELMETIA se puede restablecer si la función renal es aceptable (Ver Precauciones generales).
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Fosfato de sitagliptina: Durante los estudios clínicos controlados en sujetos sanos, dosis únicas de hasta 800 mg de sitagliptina fueron generalmente bien toleradas. En un estudio se observaron aumentos mínimos del QTc, que no se consideraron clínicamente importantes, con la dosis de 800 mg (véase Farmacocinética y farmacodinamia, Farmacodinamia, Electrofisiología cardiaca). No hay ninguna experiencia con dosis mayores de 800 mg en estudios clínicos. En estudios de Fase I de dosis múltiple, no se observaron reacciones adversas clínicas relacionadas con el medicamento con dosis de sitagliptina de hasta 600 mg por día durante periodos de hasta 10 días y 400 mg al día por periodos de hasta 28 días. Si ocurre una sobredosis, es razonable emplear las medidas de soporte usuales, como retirar del tracto digestivo el medicamento no absorbido, aplicar monitoreo clínico (incluyendo electrocardiograma) y establecer tratamiento de sostén si es necesario. La sitagliptina es poco dializable. En los estudios clínicos se extrajo aproximadamente 13.5% de la dosis durante una sesión de hemodiálisis de tres a cuatro horas. Se puede considerar hemodiálisis prolongada si es clínicamente apropiada. No se sabe si la sitagliptina es dializable por diálisis peritoneal. Clorhidrato de metformina: Se han reportado sobredosis de clorhidrato de metformina, incluyendo la ingestión de más de 50 gramos. Hubo hipoglucemia en aproximadamente 10% de los casos, pero no se ha determinado ninguna relación causal con el clorhidrato de metformina. Se ha reportado acidosis láctica en aproximadamente 32% de los casos de sobredosificación de metformina (véase Precauciones generales, Clorhidrato de metformina). La metformina es dializable, con una depuración de hasta 170 mL/min en buenas condiciones hemodinámicas, por lo que la hemodiálisis puede ser útil para extraer el medicamento acumulado en los pacientes en los que se sospecha una sobredosificación de metformina.
Presentaciones: Caja de cartón con 14, 28 o 56 comprimidos de 50 mg/500 mg, 50 mg/850 mg o 50 mg/1000 mg. Nota: No todas las presentaciones podrían estar disponibles en el mercado.
Recomendaciones sobre almacenamiento: Consérvese a no más de 25°C. Consérvese la caja bien cerrada. Protéjase de la luz.
Leyendas de protección: Su venta requiere receta médica. Prohibida la venta fraccionada del producto. No se deje al alcance de los niños. No se use durante el embarazo ni lactancia. Literatura exclusiva para Médicos. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
Nombre y domicilio del laboratorio: Undra, S.A. de C.V. Maíz No. 225, Col. Xaltocan, C.P. 16090, Deleg. Xochimilco, Ciudad de México, México. Distribuido por: LABORATORIOS LIOMONT, S.A. de C.V. Adolfo López Mateos Núm. 68, Col. Cuajimalpa, C.P. 05000, Deleg. Cuajimalpa de Morelos, México, D.F.
Número de registro del medicamento: 083M2013, SSA IV
Clave de IPPA: 0431A-MEX-2017-015342


 Cambiar de país
Cambiar de país