VIREAD®
STENDHAL
Denominación genérica: VIREAD® es la marca del tenofovir disoproxil fumarato.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: tenofovir disoproxil fumarato 300 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: El VIREAD® está indicado, en combinación con otros agentes antirretrovirales, para el tratamiento de la infección por el VIH-1. La siguiente información adicional respecto del uso de VIREAD® para el tratamiento de la infección por VIH-1 es importante: el VIREAD® no debe ser utilizado en combinación con el medicamento denominado TRUVADA® ni con ATRIPLATM (una asociación de dosis fija de efavirenz, emtricitabina y tenofovir disoproxil fumarato) (véase Precauciones generales). Estudios clínicos: pacientes sin tratamiento antirretroviral previo: estudio 903: se cuenta con datos a 144 semanas de seguimiento del estudio 903, estudio doble-ciego, controlado con principio activo, multicéntrico, que compara al VIREAD® (300 mg una vez al día) administrado en combinación con lamivudina y efavirenz contra la combinación de estavudina (d4T), lamivudina y efavirenz, en 600 pacientes infectados por VIH-1 sin tratamiento antirretroviral previo. La edad media de los pacientes fue de 36 años (intervalo de 18 a 64), 74% fueron hombres: 64% caucásicos y 20% de raza negra. La media basal del número de células CD4+ fue de 279 células/mm3 (intervalo 3-956) y la mediana basal de la carga viral plasmática fue de 77.600 copias/ml (intervalo 417-5.130.000). Los pacientes fueron estratificados en función de la carga viral y del conteo basal de células CD4+. El 43% de los pacientes tuvieron carga viral basal > 100.000 copias/ml, 39% tuvieron conteos de células CD4+ < 200 células/ mm3. Los resultados hasta la 48a y la 144a semanas se presentan en la tabla 1.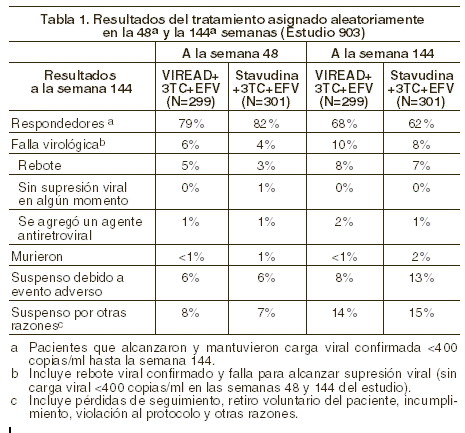
La proporción de sujetos en los que se logró reducir la carga viral a menos de 400 copias/ml tras 144 semanas de tratamiento fue similar entre los dos brazos terapéuticos para la población estratificada al inicio en base a la carga viral ( > o ≤100.000 copias/ml) o el conteo de células CD4+ ( < o ≥ 200 cels/mm3. Tras 144 semanas de tratamiento, en 62% de los pacientes tratados con VIREAD® y el 58% de los pacientes del grupo de estavudina se logró reducir de manera sostenida la carga viral a < 50 copias/ml. El incremento medio en la cuenta de células CD4+ respecto del valor basal fue de 263 cels/mm3 en el grupo de VIREAD® y de 283 cels/mm3 en el grupo de estavudina. A lo largo de las 144 semanas, 11 pacientes del grupo de VIREAD® y 9 del grupo de estavudina sufrieron un nuevo evento asociado a SIDA clase C de los CDC. Estudio 934: se han reportado los resultados a 144 semanas del estudio 934, un estudio aleatorizado, abierto, controlado con principio activo y multicéntrico en el que se compara a la combinación de VIREAD® + Emtriva® + efavirenz contra la de zidovudina + lamivudina + efavirenz en 511 pacientes infectados por el VIH-1 sin tratamiento previo. Desde la 96a a la 144ª semanas del estudio, los pacientes recibieron una asociación de dosis fija de emtricitabina + tenofovir disoproxil fumarato con efavirenz, en lugar de emtricitabina + VIREAD® con efavirenz. Los pacientes tuvieron una edad media de 38 años (intervalo 18 a 80), 86% fueron varones, 59% caucásicos y 23% de raza negra. La cuenta basal mediana de células CD4+ fue de 245 cels/mm3 (intervalo 2-1191) y la carga viral basal mediana fue de 5,01 log10 copias/ml (intervalo 3,56 a 6,54). Se estratificó a los pacientes en función de su cuenta basal de células CD4+ ( < o ≥ 200 cels/mm3); 41% de los enfermos tuvieron cuentas de células CD4+ < 200 cels/mm3, 51% tuvieron carga viral > 100.000 copias/ml. Presentamos, en la tabla 2, los resultados obtenidos a 48 y 144 semanas en aquellos pacientes que no tenían virus resistente a efavirenz al momento de iniciar el estudio: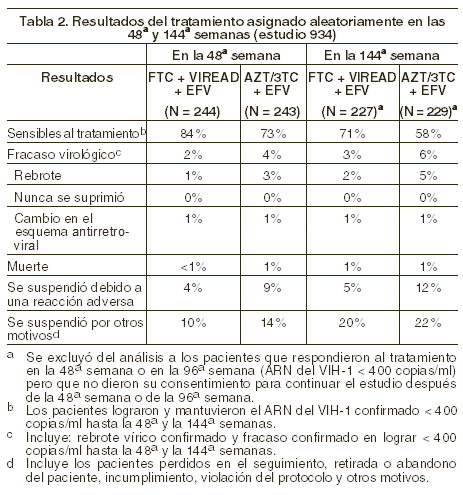
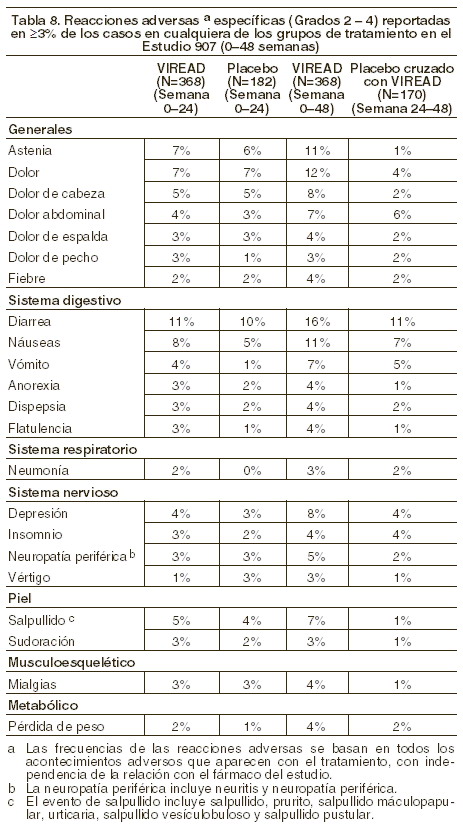
La diferencia en la proporción de pacientes que alcanzaron y mantuvieron el ARN del VIH-1 < 400 copias/ml tras las 48 semanas de tratamiento se debe en buena medida al número mayor de suspensiones del tratamiento debido a efectos adversos u otras razones observados en el grupo de pacientes tratados con ZDV/3TC. Adicionalmente, 80% de los pacientes tratados con VIREAD® y 70% de los tratados con zidovudina/lamivudina alcanzaron y mantuvieron el ARN del VIH-1 < 50 copias/ml hasta la 48ª semana (hasta 144ª semana: 64% y 56% respectivamente) En la 48ª semana, el incremento medio en la cuenta de células CD4+ con respecto al valor basal fue de 190 cels/mm3 en el grupo de EMTRIVA® + VIREAD y de 158 cels/mm3 en el grupo de zidovudina y lamivudina (en la 144ª semana: 312 y 271 cels/mm3, respectivamente). Hasta la semana 48, 7 pacientes del grupo VIREAD® + Emtriva® y 5 del grupo zidovudina y lamivudina presentaron un nuevo episodio de un evento asociado a SIDA clase C de los CDC (10 y 6 pacientes, respectivamente, hasta las 144 semanas). Pacientes tratados previamente con antirretrovirales: el estudio 907 fue un estudio multicéntrico, doble ciego, controlado con placebo, de 24 semanas de duración, en el cual se incluyó a 550 pacientes ya previamente tratados con un esquema base de medicamentos antirretrovirales, al cual se agregó VIREAD® o un placebo. Después de 24 semanas de tratamiento ciego, a todos los pacientes que continuaban en el estudio se les ofreció VIREAD® durante 24 semanas más. En general, los pacientes tuvieron un conteo medio basal plasmático de células CD4+ de 427 células/mm3 (intervalo 23-1385), el nivel basal mediano plasmático de carga viral fue de 2340 (intervalo 50-75.000) copias/ml, y la duración media del tratamiento previo con antirretrovirales fue de 5,4 años. La edad promedio de los pacientes fue de 42 años; 85% eran hombres: 69% caucásicos, 17% de color y 12% hispanos. En la figura 1 se presentan los cambios, a partir del valor basal, observados en el log10 de la carga viral plasmática durante las 48 semanas del estudio.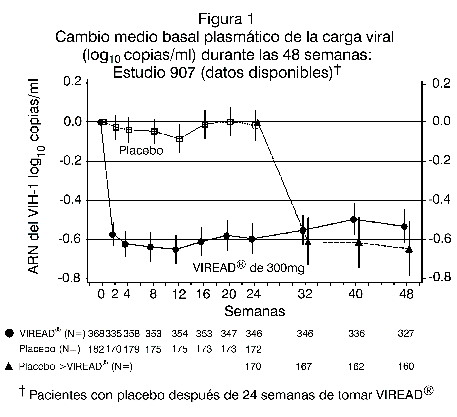
En la tabla 3 se resume el porcentaje de pacientes con carga viral < 400 copias/ml y los resultados de los pacientes a lo largo de las 48 semanas.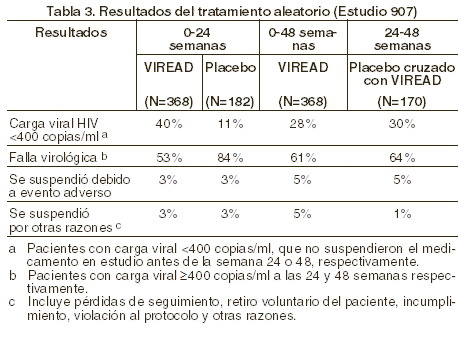
Tras 24 semanas de estudio, la proporción de pacientes con carga viral < 50 copias/ml fue mayor en el grupo que recibió VIREAD® que en el que recibió placebo (19% y 1%, respectivamente). El cambio medio en el conteo de células CD4+ a la semana 24 fue de +11 células/ mm3 en el grupo de VIREAD® y -5 células/mm3 en el grupo con placebo. En la semana 48, el cambio medio en el conteo de células CD4+ en el total de pacientes que recibieron VIREAD® fue de +4 células/mm3. Hasta la semana 24, un paciente del grupo de VIREAD® y ningún paciente del grupo de placebo experimentaron un nuevo evento adverso clase C, de acuerdo con la clasificación de los CDC.
Farmacocinética y farmacodinamia: VIREAD® es la marca registrada del tenofovir disoproxil fumarato (promedicamento del tenofovir), que es una sal de ácido fumárico de éster bis-isopropoxicarboniloximetilo derivativo de tenofovir. En vivo, el tenofovir disoproxil fumarato es convertido en tenofovir, fosfonato nucleósido acíclico (nucleótido) analógo del adenosin-5'-monofosfato. El tenofovir muestra actividad contra la transcriptasa reversa del VIH-1. El nombre químico del tenofovir disoproxil fumarato es 9-[(R)-2-[[bis[[(isopropoxicarbonilo)oxi]metoxi]fosfinil]metoxi]propil] fumarato de adenina (1:1). Su fórmula molecular es C19H30N5O10P. C4H4O4 y su peso molecular es 635,52. Su fórmula estructural es la siguiente:
El tenofovir disoproxil fumarato es un polvo cristalino, entre blanco y blancuzco, con solubilidad de 13,4 mg/ml en agua destilada a 25°C. Tiene un coeficiente de partición (log p) con buffer de octanol/fosfato (pH 6,5) de 1,25 a 25°C. Las tabletas de VIREAD® se administran por vía oral. Cada tableta contiene 300 mg de tenofovir disoproxil fumarato, equivalente a 245 mg de tenofovir disoproxil y los siguientes ingredientes inactivos: croscarmelosa de sodio, lactosa monohidratada, estearato de magnesio, celulosa microcristalina y almidón pregelatinizado. Las tabletas están recubiertas con Opadry II Y-30-10671-A, que contiene laca alumínica FD&C azul #2, hidroxipropil metilcelulosa 2910, lactosa monohidratada, dióxido de titanio y triacetina. En este documento, a reserva de que se indique lo contrario, todas las dosificaciones se refieren a tenofovir disoproxil fumarato. Microbiología: el tenofovir disoproxil fumarato es un fármaco antivírico. Mecanismo de acción: el tenofovir disoproxil fumarato es un nucléosido acíclico diéster fosfonato análogo del monofosfato de adenosina. El tenofovir disoproxil fumarato requiere de hidrólisis diéster inicial para su conversión a tenofovir y fosforilaciones subsecuentes por enzimas celulares para formar el difosfato de tenofovir. El difosfato de tenofovir inhibe la actividad de la transcriptasa reversa del VIH-1 al competir con el sustrato natural deoxiadenosin -5'-trifosfato y, una vez incorporado al ADN, por la terminación de la cadena del ADN. El difosfato de tenofovir es un inhibidor débil de las ADN-polimerasas a, b y de la ADN-polimerasa mitocondrial c en mamíferos. Actividad antiviral: la actividad antiviral in vitro de tenofovir contra aislados clínicos y de laboratorio del VIH-1 se evaluó en células linfoblastoides, en monocitos/macrófagos primarios y en linfocitos de sangre periférica. Los valores EC50 (concentración efectiva al 50%) del tenofovir estuvieron en el rango de 0,04 mM a 8,5 mM. En estudios en que se lo combinó con otros antirretrovirales, se observaron efectos entre aditivos y sinergistas del tenofovir administrado junto con: inhibidores nucleósidos de la transcriptasa reversa (abacavir, didanosina, lamivudina, stavudina, zalcitabina, zidovudina); inhibidores no-nucleósidos de la transcriptasa reversa (delavirdina, efavirenz, nevirapina); e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). El tenofovir mostró actividad antiviral in vitro contra el VIH-1 subtipos A, B, C, D, E, F, G y O (los valores EC50 fluctuaron entre 0,5 mM y 2,2 mM), así como actividad antiviral cepa-específica contra el VIH-2 (EC50 entre 1,6 mM y 4,9 mM). Resistencia: se han logrado seleccionar, en cultivo celular, cepas de VIH-1 con susceptibilidad a tenofovir reducida. Estas cepas mostraron sustitución de tipo K65R en la transcriptasa reversa y mostraron reducción de 2-4 veces en la susceptibilidad al tenofovir. En el estudio 903, realizado en pacientes sin tratamiento antirretroviral previo (VIREAD® + olamivudina + efavirenz vs. estavudina + lamivudina + efavirenz), (véase Indicaciones terapéuticas, Estudios clínicos), el análisis genotípico de virus aislado de pacientes que tuvieron falla virológica a lo largo de las 144 semanas de seguimiento mostró que las sustituciones asociadas con resistencia a efavirenz y lamivudina ocurrieron con mayor frecuencia, y sin diferencia entre los dos grupos de tratamiento. La sustitución K65R ocurrió en 8/47 (17%) de los aislamientos obtenidos de pacientes en el grupo de VIREAD® y en 2/49 (4%) de los obtenidos de los pacientes en el grupo con estavudina. De los 8 casos en los que el virus desarrolló la sustitución K65R en los pacientes tratados con VIREAD®, en 7 de los casos se presentó durante las primeras 48 semanas de tratamiento, y en uno a la semana 96. En este estudio no se identificaron otras sustituciones que confirieran resistencia al VIREAD®. En el estudio 934, también en pacientes sin tratamiento previo (VIREAD® + Emtriva® + efavirenz vs. zidovudina (AZT) /lamivudina (3TC) + efavirenz), (véase Indicaciones terapéuticas, Estudios clínicos), el análisis genotípico de los aislamientos obtenidos de todos los pacientes con carga viral > 400 copias/ml en la semana 144 o con suspensión temprana del tratamiento mostró que las sustituciones asociadas con resistencia al efavirenz fueron las más frecuentes, y se desarrollaron por igual en ambos grupos. La sustitución M184V, asociada con resistencia al EMTRIVA® y a la lamivudina, se encontró en 2/19 de los aislamientos obtenidos de pacientes en el grupo con VIREAD® + Emtriva®, y en 10/29 de los asilamientos obtenidos de los pacientes con zidovudina/lamivudina. Hasta la semana 144 de tratamiento del estudio 934, no se encontró ningún aislamiento viral en que se hubiera desarrollado la sustitución K65R en su VIH-1, esto según análisis genotípicos convencionales. Resistencia cruzada: se ha documentado que existe resistencia cruzada entre ciertos inhibidores de la transcriptasa reversa. La mutación K65R inducida por el tenofovir también se ha observado en algunos sujetos infectados con VIH-1 tratados con abacavir, didanosina o zalcitabina. Las cepas de VIH-1 que expresan esta mutación muestran también susceptibilidad reducida a emtricitabina y a lamivudina. Por consiguiente, la resistencia cruzada entre estos medicamentos puede ocurrir en pacientes cuyo virus albergue la mutación K65R. En cepas clínicas de VIH-1 (N=20) con media de 3 sustituciones de la transcriptasa reversa asociadas a zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N), se encontró disminución de 3,1 veces en la susceptibilidad al tenofovir. El VIH-1 multirresistente a nucleósidos que expresa una mutación de inserción doble T69S en la transcriptasa reversa también muestra susceptibilidad reducida al tenofovir. En los estudios 902 y 907, conducidos en pacientes con tratamiento antirretroviral previo (VIREAD® + tratamiento basal estándar, o TBE vs. Placebo + TBE), (véase Indicaciones terapéuticas, Estudios clínicos), se obtuvieron aislamientos virales en 14/304 (5%) de los pacientes tratados con VIREAD® que tuvieron falla virológica a la semana 96 con susceptibilidad reducida al tenofovir > 1,4 veces (mediana, 2,7 veces). El análisis genotípico comparativo de los aislamientos obtenidos al inicio del estudio y tras la falla virológica mostraron el desarrollo de la mutación K65R en el gen de la transcriptasa reversa del VIH-1. La respuesta virológica a la terapia con VIREAD® se evaluó con respecto al genotipo viral basal (N=222) en pacientes con tratamiento antirretroviral previo que participaron en los estudios 902 y 907. En estos estudios clínicos, 94% de los participantes evaluados tuvieron en el punto basal cepas de VIH-1 que expresaban por lo menos una mutación que confiere resistencia a inhibidores de transcriptasa reversa análogos de nucleótidos (ITRAN), incluyendo mutaciones asociadas con resistencia a zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N), la sustitución asociada con resistencia a abacavir/emtricitabina/lamivudina (M184V) y otras. Además, la mayoría de los participantes evaluados tuvieron mutaciones asociadas a resistencia a los inhibidores de proteasa (IP) o de inhibidores de transcriptasa reversa no nucleótidos (ITRNN). Las respuestas virológicas de los pacientes en el subestudio genotípico fueron similares a los resultados del grupo general. Se realizaron varios análisis exploratorios para evaluar el efecto de sustituciones específicas y los patrones de sustitución en el resultado virológico de los pacientes tratados con VIREAD®. Se observaron grados variables de resistencia cruzada a VIREAD® en cepas con sustituciones preexistentes de resistencia a zidovudina; el grado de resistencia pareció depender del número de mutaciones específicas. Los pacientes tratados con VIREAD® cuyo VIH-1 expresó 3 o más mutaciones asociadas a resistencia a zidovudina que incluían ya sea la mutación M41L o la L210W de la transcriptasa reversa mostraron respuestas reducidas a la terapia con VIREAD®; sin embargo, estas respuestas fueron mayores a las obtenidas con placebo. La presencia de la mutación D67N, K70R, T215Y/F o K219Q/E/N al parecer no afectó las respuestas a la terapia con VIREAD®. En el análisis preestablecido en el protocolo, la respuesta virológica al VIREAD® no se redujo en pacientes con virus que tuvieran la mutación M184V, asociada con resistencia a lamivudina/emtricitabina/abacavir. En presencia de mutaciones asociadas a resistencia a la zidovudina, la mutación M184V no afectó las respuestas medias de la carga viral en pacientes tratados con VIREAD®. La respuesta de la carga viral del VIH-1 en estos pacientes fue consistente hasta la semana 48. Análisis fenotípico de aislamientos obtenidos en los estudios 902 y 907: la respuesta virológica a la terapia con VIREAD® se evaluó con respecto al fenotipo basal (N=100) en pacientes que ya habían recibido antirretrovirales y que participaron en dos estudios controlados. El análisis fenotípico basal del VIH-1 en los pacientes de estos estudios mostró una correlación entre la susceptibilidad basal a VIREAD® y la respuesta a la terapia con este medicamento. En la tabla 4 se resume la respuesta de la carga viral en función de la susceptibilidad basal al VIREAD®.
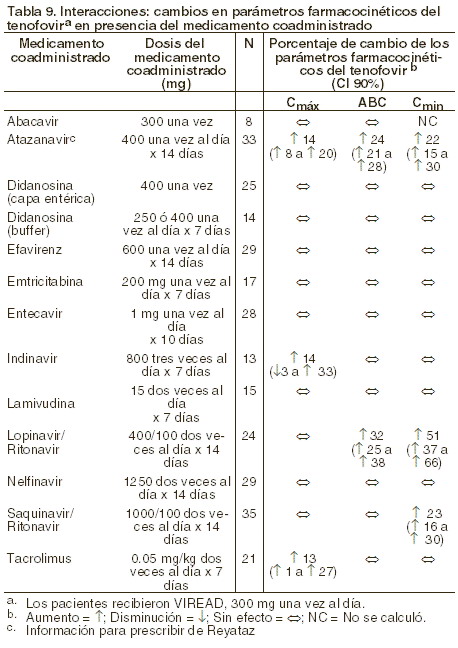
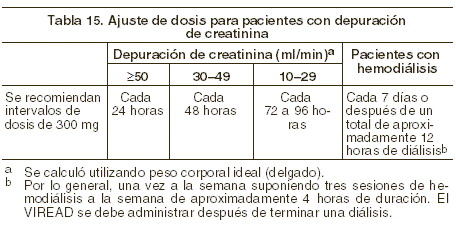
Farmacología clínica: farmacocinética: los parámetros farmacocinéticos del tenofovir disoproxil fumarato han sido evaluados en voluntarios sanos y en individuos infectados por el VIH-1. No se encontraron diferencias en la farmacocinética del tenofovir entre estas dos poblaciones. Absorción: el VIREAD® es una prodroga diéster hidrosoluble del ingrediente activo tenofovir. En pacientes en ayuno, la biodisponibilidad oral de tenofovir es de aproximadamente 25%. Después de la administración oral de una sola dosis de VIREAD® de 300 mg a pacientes infectados con VIH-1 en ayuno, se alcanzó la concentración máxima en suero (Cmáx) en 1,0 ± 0,4 hrs. Los valores de Cmáx y ABC son de 0,30 ± 0,30 mg/ml y 2,29 ± 0,69mg*h/ml respectivamente. La farmacocinética del tenofovir es proporcional a la dosis en un rango entre 75 y 600 mg, y no se ve afectada por dosis repetidas. Distribución: la unión in vitro del tenofovir con el plasma humano o con proteínas de suero es menor a 0,7 y 7,2%, respectivamente, cuando la concentración del tenofovir está en un intervalo de 0,01 y 25 g/ml. Tras la administración intravenosa de 1,0 mg/kg y 3,0 mg/kg de tenofovir, el volumen de distribución en estado estable es de 1,3 ± 0,6 l/kg y 1,2 ± 0,4 l/kg respectivamente. Metabolismo y eliminación: los estudios in vitro indican que ni el tenofovir disoproxil ni el tenofovir son substratos de las enzimas del complejo CYP. Tras la administración IV de tenofovir, de 70 a 80% de la dosis se recupera sin cambios en la orina tras las primeras 72 horas. La vida media de eliminación terminal del tenofovir es de aproximadamente 17 horas tras la administración de una dosis única por vía oral. Después de dosis orales múltiples de VIREAD®, a razón de 300 mg una vez al día (después del alimento), 32 ± 10% de la dosis administrada se recupera en la orina después de 24 horas. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Puede haber competencia por la eliminación con otros compuestos que también se eliminan por vía renal. Efectos del alimento en la absorción oral: la ingesta de VIREAD® después de una comida rica en grasa (<126>700 a 1.000 kcal que contenga de 49 a 50% de grasa) aumenta la biodisponibilidad oral, con incremento en el ABC0-∞ del tenofovir de aproximadamente 40% e incremento en la Cmáx de aproximadamente 14%. Sin embargo, la coadministración de VIREAD® con una comida ligera no tuvo efecto importante en la farmacocinética del tenofovir en comparación con la observada al administrar el medicamento en ayunas. La comida retrasa el tiempo de la Cmáx del tenofovir aproximadamente en 1 hora. Cuando el contenido de la dieta no se controló, la Cmáx y la ABC del tenofovir fueron 0,33 ± 0,12 mg/ml y 3,32 ± 1,37 mg*h/ml, respectivamente, después de dosis múltiples de VIREAD® de 300 mg una vez al día, administradas después de consumir alimentos. Poblaciones especiales: raza: hasta ahora, el número de pacientes de raza diferente de la caucásica que ha recibido tenofovir es demasiado pequeño como para determinar si existen diferencias interraciales en la farmacocinética del medicamento. Sexo: los parámetros farmacocinéticos del tenofovir son similares entre pacientes masculinos y femeninos. Pacientes pediátricos y geriátricos: no se han realizado estudios farmacocinéticos en niños ( < 18 años) o en personas mayores ( > 65 años). Pacientes con disfunción renal: la farmacocinética del tenofovir sí se altera en pacientes con falla renal (véase Precauciones generales). En pacientes con depuración de creatinina < 50 ml/min o con enfermedad renal en fase terminal (ERFT) que requiere diálisis, se incrementan la Cmáx y el ABC0-∞ del tenofovir (tabla 5). Se recomienda que se modifique el intervalo de administración de VIREAD® en pacientes con depuración de creatinina < 50 ml/min o en pacientes con ERFT que requieran diálisis (véase Dosis y vía de administración).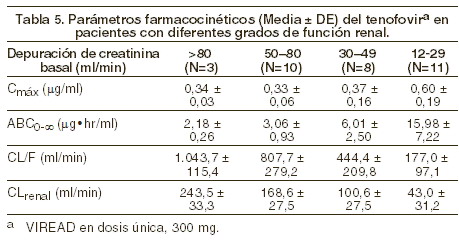
El tenofovir es eficientemente removido por hemodiálisis, con un coeficiente de extracción cercano al 54%. Después de la administración de una dosis única de 300 mg de VIREAD®, una sesión de hemodiálisis de 4 horas removió aproximadamente el 10% de la dosis de tenofovir administrada. Pacientes con disfunción hepática: se han estudiado las propiedades farmacocinética del tenofovir después una dosis única de 300 mg de VIREAD®, en pacientes no infectados por el VIH y con disfunción hepática moderada a grave. No hubo alteraciones importantes en las propiedades farmacocinéticas del tenofovir en pacientes con disfunción hepática, en comparación con los pacientes con función hepática normal. No se requiere ningún cambio de la dosificación de VIREAD® en pacientes con disfunción hepática.
Contraindicaciones: El VIREAD® está contraindicado en pacientes con hipersensibilidad previamente demostrada a cualquiera de los componentes del producto.
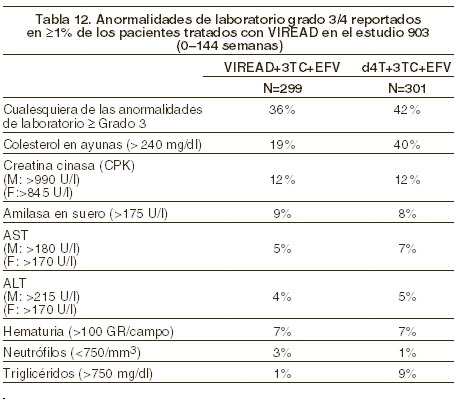
Precauciones generales: Acidosis láctica/hepatomegalia grave con esteatosis: se han reportado casos de acidosis láctica y hepatomegalia grave con esteatosis, incluso fatales, en pacientes que reciben análogos nucleósidos solos o en combinación con otros agentes antirretrovirales. La mayoría de los casos se han reportado en mujeres. La obesidad y la exposición prolongada a los análogos nucleósidos parecen ser factores de riesgo. Debe ejercerse particular precaución al prescribir análogos nucleósidos a personas con factores de riesgo identificados para hepatopatía, sin dejar de tomar en cuenta que se han reportado casos aun en personas sin ningún factor de riesgo identificable. Debe suspenderse el tratamiento con VIREAD® en cualquier paciente que desarrolla datos clínicos o de laboratorio sugestivos de acidosis láctica o de hepatotoxicidad significativa (incluyendo hepatomegalia y esteatosis aún sin elevación concomitante de las transaminasas). Exacerbación de la hepatitis después de la suspensión del tratamiento: se han reportado exacerbaciones agudas graves de la hepatitis en pacientes coinfectados con el VIH-1 y el VBH que suspenden el tratamiento con VIREAD. Se debe hacer un monitoreo estricto con análisis clínicos y de laboratorio a los pacientes que tienen una infección concomitante por el VIH-1 y el VHB y que suspenden VIREAD, hasta algunos meses después de interrumpir el tratamiento. Si procede, puede estar justificado reanudar el tratamiento de la hepatitis B. En los pacientes con enfermedad hepática avanzada o cirrosis, no se recomienda la suspensión del tratamiento de la hepatitis B, ya que la exacerbación de la hepatitis después del tratamiento puede causar una descompensación hepática. Nueva aparición o empeoramiento de la disfunción renal: el tenofovir se elimina principalmente por los riñones. Se han notificado casos de trastornos renales, entre ellos casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia severa), asociados con el uso de VIREAD® (véase Reacciones secundarias y adversas, Experiencia posterior a la comercialización). Se recomienda calcular la depuración de creatinina en todos los pacientes antes de iniciar la terapia y según se requiera clínicamente durante el tratamiento con VIREAD®. Debe realizarse la vigilancia sistemática de la depuración de creatinina calculada y del fósforo sérico en todos los pacientes con riesgo de disfunción renal, incluidos los pacientes que han sufrido anteriormente trastornos renales cuando recibian HEPSERA (dipivoxilo de adefovir). Se recomienda el ajuste del intervalo de dosificación de VIREAD® y la vigilancia estricta de la función renal en todos los pacientes con una depuración de creatinina < 50 ml/ min (véase Dosis y vía de administración). No hay información sobre la inocuidad ni la eficacia en los pacientes con disfunción renal que hayan recibido VIREAD® siguiendo estas pautas de dosificación, por lo que el beneficio potencial del tratamiento con VIREAD® debe evaluarse en relación con el posible riesgo de la toxicidad renal. Se debe evitar el uso concurrente de VIREAD® con el uso reciente o concomitante de un medicamento nefrotóxico. Administración concomitante de productos: VIREAD® no debe utilizarse en asociación con los productos de asociación de dosis fijas TRUVADA® o ATRIPLATM, ya que el tenofovir disoproxil fumarato es un componente de estos productos. VIREAD® no debe administrarse en combinación con HEPSERA® (dipivoxilo de adefovir). Pacientes con infección concomitante por el VIH-1 y el VHB: antes de iniciar el tratamiento con VIREAD®, se recomienda analizar la presencia de hepatitis B crónica en todos los pacientes infectados por el VIH-1. Disminución de la densidad ósea: se debe planear la vigilancia de la densidad ósea (DMO)para los pacientes infectados por el VIH-1 que tienen antecedentes de fracturas óseas patológicas o con riesgo de osteopenia. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos para todos los pacientes. Se debe obtener asesoramiento adecuado si se sospecha de la presencia de anomalías óseas. En pacientes infectados por el VIH-1 y tratados con VIREAD®, en el estudio 903, tras 144 semanas, se observó disminución de la densidad mineral ósea (DMO) en columna lumbar y cadera en ambos grupos de estudio. El porcentaje de reducción en la DMO (media ± DE) de la columna lumbar a las 144 semanas fue significativamente mayor en el grupo tratado con VIREAD® + lamivudina + efavirenz (-2,2% ± 3,9) en comparación con los pacientes que recibieron stavudina + lamivudina + efavirenz (-1,0 ± 4,6). Los cambios en la DMO a nivel de cadera fueron similares en ambos grupos (-2,8 ± 3,5 en el grupo de VIREAD® vs. -2,4 ± 4,5 en el grupo de estavudina). En ambos grupos, la mayor parte de la reducción en la DMO ocurrió en las primeras 24-48 semanas del estudio, manteniéndose estable desde ese momento hasta la semana 144. El porcentaje de pacientes que sufrieron una reducción de al menos en 5% en la DMO lumbar o del 7% en la cadera fue del 28% en el grupo de VIREAD® y de 21% en el grupo de estavudina. Se reportaron fracturas clínicamente relevantes (excluyendo dedos de las manos o pies) en 4 pacientes del grupo de VIREAD® y en 6 pacientes del grupo de estavudina. Además, hubo aumentos significativos en los niveles de cuatro marcadores bioquímicos del metabolismo óseo (fosfatasa alcalina ósea sérica, osteocalcina sérica, telopéptido C sérico y telopéptido N urinario) en el grupo VIREAD® con relación al grupo de estavudina, lo que sugiere mayor resorción ósea. Los niveles séricos medios de hormona paratiroidea y de vitamina D 1,25, también fueron más altos en el grupo que recibió VIREAD®. Excepto por la fosfatasa alcalina ósea, los valores de estos marcadores se mantuvieron dentro del rango normal. Se desconoce el impacto real sobre la salud ósea y el riesgo de fracturas que tienen los cambios en la DMO y en los marcadores bioquímicos de resorción ósea asociados con el uso de VIREAD®. Se han notificado casos de osteomalacia (asociada a tubulopatía renal proximal) en relación con el uso de VIREAD® (véase Reacciones secundarias y adversas). Redistribución de la grasa corporal: en los pacientes infectados por el VIH-1, se han descrito eventos de redistribución/acumulación de grasa corporal, incluyendo obesidad central; hiperplasia de grasa dorsocervical (joroba de búfalo), emaciación periférica o facial, agrandamiento de mamas y "aspecto cushingoide" en pacientes tratados con antirretrovirales en asociación. El mecanismo y las consecuencias a largo plazo de estos eventos se desconocen actualmente. No se ha establecido una relación causal. Síndrome de reconstitución inmunológica: se han reportado casos de este síndrome en pacientes infectados por el VIH que reciben medicamentos antirretrovirales, incluyendo al VIREAD®. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyo sistema inmune se recupera pueden desarrollar una respuesta inflamatoria a infecciones oportunistas subyacentes o residuales por agentes como el Mycobacterium avium, Citomegalovirus, Pneumocystis jirovecii (PCP) o el bacilo de la tuberculosis, que pueden ameritar de evaluación y tratamiento específicos. Fracaso virológico: los estudios clínicos en los pacientes infectados por el VIH han demostrado que algunas pautas que sólo contienen tres inhibidores nucleósidos de la retrotranscriptasa (INRT) son generalmente menos eficaces que las pautas de tres fármacos que contienen dos INRT asociados a un inhibidor no nucleósido de la retrotranscriptasa o a un inhibidor de la proteasa del VIH-1. En particular, se han notificado el fracaso virológico temprano y tasas altas de sustituciones de resistencia. Por lo tanto, las pautas con tres nucleosídicos deben emplearse con precaución. Se debe monitorear atentamente a los pacientes que sólo reciban una pauta con tres nucleosídicos y se debe estudiar la modificación de su tratamiento. Toxicología animal o farmacología: los estudios de toxicología animal realizados con el tenofovir y el tenofovir disoproxil fumarato en ratas, perros y monos sometidos a concentraciones (basados en las AUC) al menos 6 veces mayores a las observadas en humanos mostraron toxicidad ósea. En los monos la toxicidad ósea se tradujo en osteomalacia. La osteomalacia que se observó en los monos mostró ser reversible al reducir la dosis o al discontinuar el tenofovir. En ratas y perros, la toxicidad ósea se manifestó como densidad ósea reducida. Se desconoce el mecanismo(s) implícito(s) en la toxicidad ósea. En 4 especies animales se observó evidencia de toxicidad renal. En diferentes grados, se observó incremento en la creatinina sérica, BUN, glucosuria, proteinuria, fosfaturia y/o calciuria y disminución en el fosfato sérico en estos animales. Estas toxicidades se observaron en exposiciones (basados en las AUC) de 2-20 veces superiores a las observadas en humanos. Se desconoce la relación de las anormalidades renales, en particular la de fosfaturia, con la toxicidad ósea. Uso pediátrico: no se ha establecido la seguridad y la efectividad en pacientes menores a 18 años de edad. Uso geriátrico: los estudios clínicos con VIREAD® no incluyeron un número suficiente de pacientes con edad igual o superior a los 65 años como para poder determinar que tienen respuestas al medicamento diferentes a las de sujetos más jóvenes. Como regla general, la dosificación en pacientes de la tercera edad debe hacerse con precaución, tomando en cuenta la mayor incidencia de disfunción hepática, renal o cardíaca, así como de patologías concomitantes y de la ingesta de otro tipo de medicamentos. Información para los pacientes: se debe informar a los pacientes lo siguiente: VIREAD® no cura la infección por el VIH-1 y los pacientes pueden seguir presentando enfermedades asociadas con esta infección, incluidas las infecciones oportunistas. Los pacientes deben permanecer bajo el cuidado de un médico cuando utilizan VIREAD®. No se ha comprobado que el uso de VIREAD® reduzca el riesgo de transmisión de la infección por el VIH-1 a otras personas por medio del contacto sexual o de la contaminación de sangre. Se desconocen los efectos a largo plazo de VIREAD®. Las tabletas de VIREAD® son sólo para administración por vía oral. No debe suspenderse la administración de VIREAD® sin informar primero a su médico. Es importante tomar VIREAD® en un tratamiento de asociación, con un intervalo de dosificación regular, para evitar que el paciente olvide tomar la dosis. Se ha notificado la aparición de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales. El tratamiento con VIREAD® deberá interrumpirse en cualquier paciente que presente síntomas clínicos que indiquen la presencia de acidosis láctica o de hepatotoxicidad pronunciada (incluso náuseas, vómitos, molestias gástricas poco habituales o inesperadas y debilidad) (véase Precauciones generales). Se debe realizar a los pacientes con infección por el VIH-1 la prueba para detectar la presencia del virus de la hepatitis B (VIH) antes de iniciar el tratamiento antirretroviral (véase Precauciones generales). Se han notificado exacerbaciones agudas y graves de la hepatitis B en los pacientes infectados concomitantemente por el VIH-1 y el VHB que suspendieron la administración de VIREAD® (véase Precauciones generales). Se han notificado casos de disfunción renal, entre ellos casos de insuficiencia renal aguda y del síndrome de Fanconi. Se debe evitar el uso de VIREAD® con el uso reciente o concomitante de un medicamento nefrotóxico (véase Precauciones generales). Puede ser necesario justar el intervalo de dosificación de VIREAD® en los pacientes con disfunción renal (véase Dosis y vía de administración). VIREAD® no debe utilizarse en asociación con los productos de asociación de dosis fijas TRUVADA® o ATRIPLATM, ya que es un componente de dichos productos (véase Precauciones generales). VIREAD® no debe administrarse en combinación con HEPSERA® (véase Precauciones generales). Se ha observado disminución de la densidad ósea con el uso de VIREAD®. Se debe plantear la vigilancia de la densidad ósea en los pacientes que tengan antecedentes de fracturas óseas patológicas o con riesgo de osteopenia (véase Precauciones generales).
Restricciones de uso durante el embarazo y la lactancia: Embarazo: embarazo categoría B: se realizaron estudios de reproducción en ratas y conejos con dosis 14 y 19 veces la dosis humana basada en la comparación de la superficie corporal y no se encontró ninguna evidencia de alteración en la fertilidad o daños al feto por el tenofovir. Sin embargo, no hay estudios adecuados y bien controlados con mujeres embarazadas. Como los estudios de reproducción animal no siempre son predictivos de la respuesta humana, el VIREAD® sólo debe usarse durante el embarazo si es muy necesario. Registro de embarazos con antirretrovirales: se invita a los profesionales médicos a que registren a las pacientes que se queden embarazadas en el siguiente sitio de internet: www.kendle.com/registries. Madres lactando: los centros de control y prevención de enfermedades recomiendan que las madres infectadas con VIH no den pecho a sus hijos para evitar el riesgo de transmisión postnatal del VIH. Estudios con ratas han demostrado que el tenofovir es secretado en la leche. Se desconoce si el tenofovir se excreta en la leche humana. Debido tanto al riesgo de transmisión del VIH como al potencial de reacciones adversas serias que puede haber en los lactantes, se debe pedir a las madres que no den pecho a sus bebés si están tomando VIREAD®.
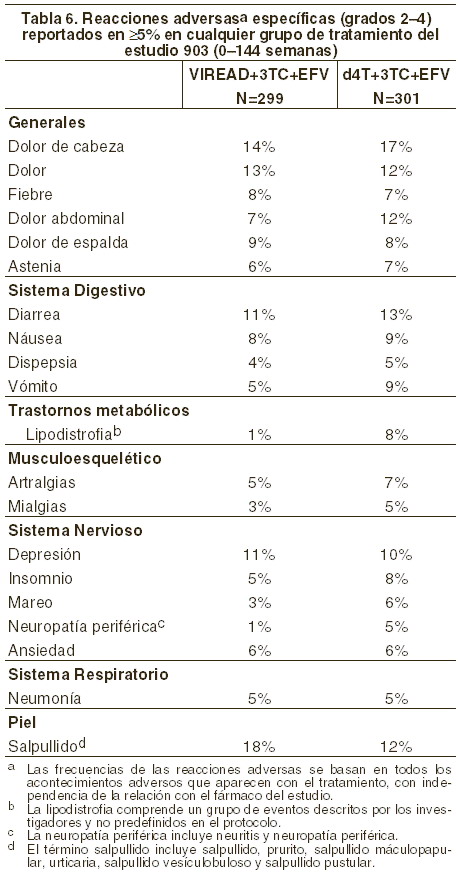
Reacciones secundarias y adversas: En otros apartados de la información para prescribir, se tratan las siguientes reacciones adversas: acidosis láctica o hepatomegalia grave con esteatosis (véase Precauciones generales). Exacerbaciones agudas y graves de la hepatitis B (véase Precauciones generales). Nueva aparición o empeoramiento de la disfunción renal (véase Precauciones generales). Disminución de la densidad ósea (véase Precauciones generales). Síndrome de reconstitución inmunológica (véase Precauciones generales). Experiencia en estudios clínicos: se han tratado más de 12.000 pacientes con VIREAD® solo o en combinación con otros productos antirretrovirales en períodos de 28 días a 215 semanas en estudios clínicos estudios de acceso expandido. Un total de 1.544 pacientes han recibido 300 mg una vez al día de VIREAD® en estudios clínicos; más de 11.000 pacientes han recibido VIREAD® en estudios de acceso expandido. Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Las reacciones adversas más frecuentes (incidencia ≥10%, grados 2-4) identificados a partir de cualquiera de los tres grandes ensayos clínicos controlados son erupción cutánea, diarrea, cefalea, dolor, depresión, astenia y náuseas. Pacientes sin tratamiento antirretroviral previo: estudio 903 - Reacciones adversas aparecidas durante el tratamiento: las reacciones adversas más frecuentemente encontradas en el estudio controlado comparativo doble-ciego en el cual 600 pacientes sin tratamiento antirretroviral previo recibieron VIREAD® (N=299) o estavudina