VYTORIN
MSD
Denominación genérica: Ezetimiba, Simvastatina
Forma farmacéutica y formulación: Formulación: Cada comprimido contiene: Ezetimiba 10 mg, Simvastatina 10 mg, 20 mg, 40 mg, 80 mg, Excipiente cbp 1 comprimido. Cada comprimido contiene los siguientes ingredientes inactivos: butilhidroxianisol, ácido cítrico monohidratado, croscarmelosa, hipromelosa, lactosa monohidratada, estearato de magnesio, celulosa microcristalina y galato de propilo
Indicaciones terapéuticas: Hipercolesterolemia primaria VYTORIN está indicado como tratamiento agregado a la dieta para disminuir las concentraciones elevadas de colesterol total, colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteínas B (Apo B), triglicéridos (TG) y colesterol de lipoproteínas de no alta densidad (C-no-HDL) y para aumentar el colesterol de lipoproteínas de alta densidad (C-HDL) en pacientes adultos y adolescentes (10 a 17 años de edad) con hipercolesterolemia primaria (heterocigótica familiar y no familiar) o con hiperlipidemia mixta. Se puede agregar fenofibrato al tratamiento con VYTORIN en pacientes adultos con hiperlipidemia mixta que requieran de una mayor reducción de triglicéridos (TG) y C-no-HDL, así como de un incremento en el C-HDL. Hipercolesterolemia familiar homocigótica VYTORIN está indicado para disminuir las concentraciones elevadas de colesterol total y de C-LDL en pacientes adultos y adolescentes (10 a 17 años de edad) con hipercolesterolemia familiar homocigótica, los cuales pueden recibir también tratamientos adjuntos (p. ej., aféresis de LDL).
Farmacocinética y farmacodinamia: CLASE TERAPÉUTICA Vytorin (ezetimiba/simvastatina) es un producto hipolipemiante que inhibe selectivamente la absorción intestinal de colesterol y esteroles vegetales relacionados, e inhibe la síntesis endógena de colesterol. ABSORCIÓN Ezetimiba Después de la administración oral, la ezetimiba se absorbe rápidamente y se conjuga ampliamente a un glucurónido fenólico farmacológicamente activo (ezetimiba-glucurónido). El promedio de las concentraciones plasmáticas máximas (Cmáx) ocurre en las siguientes 1 a 2 horas para ezetimiba- glucurónido y de 4 a 12 horas para ezetimiba. La biodisponibilidad absoluta de ezetimiba no se puede determinar ya que el compuesto es prácticamente insoluble en medios acuosos adecuados para inyección. La administración concomitante de alimentos (comidas con alto contenido de grasa o sin grasa) no tuvieron efecto sobre la biodisponibilidad oral de ezetimiba cuando se administró como tabletas de ezetimiba de 10 mg. Simvastatina La disponibilidad del b-hidroxiácido a la circulación sistémica después de una dosis oral de simvastatina resultó menor al 5% de la dosis, consistente con la extensa extracción hepática de primer paso. Los principales metabolitos de simvastatina presentes en el plasma humano son b-hidroxiácido y cuatro metabolitos activos adicionales. En relación con el estado de ayuno, los perfiles plasmáticos tanto de los inhibidores activos como de los totales no se vieron afectados cuando se administró simvastatina inmediatamente antes de una comida de prueba. DISTRIBUCIÓN Ezetimiba Ezetimiba y ezetimiba-glucurónido se unen en un 99.7% y 88 a 92% a las proteínas plasmáticas humanas, respectivamente. Simvastatina Tanto la simvastatina como el b-hidroxiácido se unen a las proteínas del plasma humano (95%). La farmacocinética de dosis únicas y múltiples de simvastatina mostró que no se produce una acumulación de fármaco después de múltiples dosis. En todos los estudios farmacocinéticos anteriores, la concentración plasmática máxima de inhibidores se produjo de 1.3 a 2.4 horas después de la dosis. METABOLISMO Ezetimiba Ezetimiba se metaboliza principalmente en el intestino delgado y el hígado a través de la conjugación con glucurónido (una reacción de fase II) con posterior excreción biliar. Se observó un metabolismo oxidativo mínimo (una reacción de fase I) en todas las especies evaluadas. Ezetimiba y ezetimiba-glucurónido son los principales compuestos derivados del fármaco detectados en el plasma, que constituyen aproximadamente el 10 a 20% y de 80 a 90% del fármaco total en plasma, respectivamente. Tanto ezetimiba como ezetimiba-glucurónido se eliminan lentamente del plasma, con evidencia de reciclado enterohepático significativo. La vida media de ezetimiba y ezetimiba-glucurónido es de aproximadamente 22 horas. Simvastatina La simvastatina es una lactona inactiva que se hidroliza rápidamente in vivo al b-hidroxiácido correspondiente, un potente inhibidor de la HMG-CoA reductasa. La hidrólisis se lleva a cabo principalmente en el hígado; la velocidad de hidrólisis en el plasma humano es muy lenta. En el hombre, simvastatina se absorbe bien y sufre una extensa extracción hepática de primer paso. La extracción en el hígado depende del flujo sanguíneo hepático. El hígado es el principal sitio de acción, con excreción posterior de equivalentes del fármaco en la bilis. En consecuencia, la disponibilidad del fármaco activo en la circulación sistémica es baja. Después de una inyección intravenosa del metabolito b-hidroxiácido, su vida media promedio es de 1.9 horas. ELIMINACIÓN Ezetimiba Tras la administración oral de 14C-ezetimiba (20 mg) a sujetos humanos, ezetimiba total representó aproximadamente el 93% de la radiactividad total en el plasma. Aproximadamente el 78% y el 11% de la radiactividad administrada se recuperó en las heces y la orina, respectivamente, durante un periodo de recolección de 10 días. Después de 48 horas, no hubo niveles detectables de radiactividad en el plasma. Simvastatina Tras una dosis oral de simvastatina radiactiva a humanos, el 13% de la radiactividad se excretó en la orina y el 60% en las heces durante las siguientes 96 horas. La cantidad recuperada en las heces representa los equivalentes del fármaco absorbido excretados en la bilis, así como el fármaco no absorbido. Después de una inyección intravenosa del metabolito b-hidroxiácido, se excretó un promedio de sólo 0.3% de la dosis IV en la orina como inhibidores. Características en Grupos de Pacientes Especiales Pacientes pediátricos La absorción y el metabolismo de ezetimiba son similares entre los niños y adolescentes (de 10 a 18 años) y los adultos. Con base en ezetimiba total, no hay diferencias farmacocinéticas entre adolescentes y adultos. No hay datos farmacocinéticos disponibles en la población pediátrica < 10 años de edad. Pacientes geriátricos Las concentraciones plasmáticas de ezetimiba total son aproximadamente 2 veces mayores en los adultos de edad avanzada (≥65 años) que en los jóvenes (18 a 45 años). La reducción de C-LDL y el perfil de seguridad son comparables entre sujetos de edad avanzada y jóvenes tratados con ezetimiba. Insuficiencia hepática Después de una dosis única de 10 mg de ezetimiba, el área bajo la curva (AUC) promedio de ezetimiba total aumentó aproximadamente 1.7 veces en pacientes con insuficiencia hepática leve (puntuación Child-Pugh de 5 o 6), en comparación con sujetos sanos. En un estudio de dosis múltiples de 14 días (10 mg al día) en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), el AUC promedio de ezetimiba total aumentó aproximadamente 4 veces el Día 1 y el Día 14, en comparación con los sujetos sanos. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve. Debido a los efectos desconocidos de una mayor exposición a ezetimiba en pacientes con insuficiencia hepática moderada o grave (puntuación Child-Pugh > 9), no se recomienda ezetimiba en estos pacientes (Ver Precauciones generales). Insuficiencia renal Ezetimiba Después de una dosis única de 10 mg de ezetimiba, en pacientes con enfermedad renal grave (n=8); (promedio de depuración de creatinina ≤30 mL/min/1.73 m2), el promedio del AUC de ezetimiba total aumentó aproximadamente 1.5 veces, en comparación con sujetos sanos (n=9). Un paciente adicional en este estudio (post trasplante renal y que recibía múltiples medicamentos, incluyendo ciclosporina) tuvo una exposición 12 veces mayor a ezetimiba total. Simvastatina En un estudio de pacientes con insuficiencia renal grave (depuración de creatinina < 30 mL/min), las concentraciones plasmáticas de inhibidores totales después de una sola dosis de un inhibidor de la HMG-CoA reductasa relacionado fueron aproximadamente dos veces mayores que las de los voluntarios sanos. Sexo Las concentraciones plasmáticas de ezetimiba total son ligeramente mayores ( < 20%) en las mujeres que en los hombres. La disminución del C-LDL y el perfil de seguridad son similares entre hombres y mujeres tratados con ezetimiba. Raza Basándose en un meta-análisis de estudios farmacocinéticos con ezetimiba, no hubo diferencias farmacocinéticas entre las personas de raza negra y las de raza blanca. Estudios Clínicos En estudios clínicos controlados, VYTORIN redujo significativamente el colesterol total (C-total), colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteína B (Apo B), triglicéridos (TG), y colesterol de lipoproteínas de no alta densidad (C-no-HDL) y aumentó el colesterol de lipoproteínas de alta densidad (C-HDL) en los pacientes con hipercolesterolemia. Hipercolesterolemia Primaria VYTORIN Se reportan cinco estudios multicéntricos, doble ciego, realizados con VYTORIN en pacientes con hipercolesterolemia primaria: dos fueron comparaciones con simvastatina, dos fueron comparaciones con atorvastatina, y uno fue una comparación con rosuvastatina. En un estudio multicéntrico, doble ciego, controlado con placebo, de 12 semanas, 887 pacientes hipercolesterolémicos fueron aleatorizados a uno de diez grupos de tratamiento: placebo, ezetimiba (10 mg), simvastatina (10 mg, 20 mg, 40 mg, u 80 mg), o ezetimiba y simvastatina coadministradas equivalente a VYTORIN (10/10, 10/20, 10/40, y 10/80). Cuando los pacientes que recibieron VYTORIN fueron comparados con los que recibieron todas las dosis de simvastatina, VYTORIN redujo significativamente el C-total, C-LDL, Apo B, TG, C-no-HDL, y la proteína C-reactiva. Los efectos de VYTORIN sobre el C-HDL fueron similares a los efectos observados con simvastatina. Un análisis posterior mostró que VYTORIN aumentó significativamente el C-HDL en comparación con el placebo. (Ver Tabla 1.)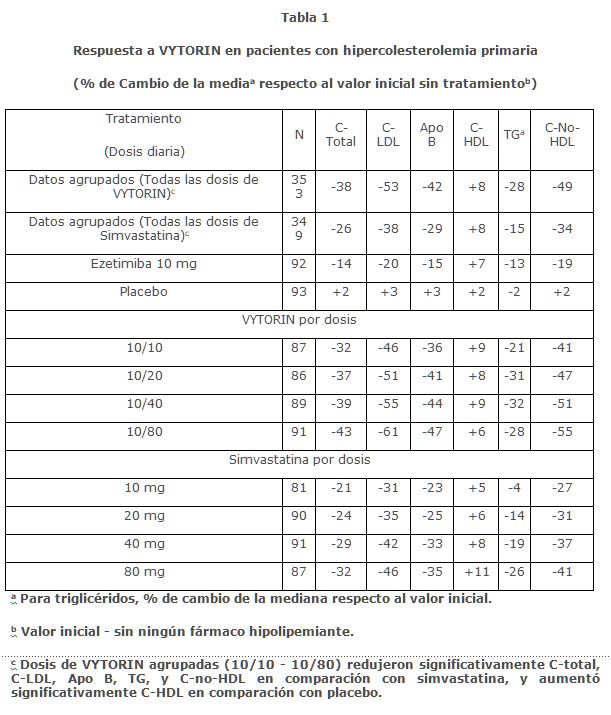
En un estudio diseñado de manera similar, los resultados para todos los parámetros lipídicos fueron generalmente consistentes. En un análisis agrupado de estos dos estudios, la respuesta de los lípidos a VYTORIN fue similar en los pacientes con niveles de TG superiores o inferiores a 200 mg/dL. En un estudio multicéntrico, doble ciego, controlado, de 23 semanas, 710 pacientes con cardiopatía coronaria conocida o equivalentes de riesgo de cardiopatía coronaria, según se definen en los lineamientos del NCEP ATP III, y un C-LDL ≥130 mg/dL fueron aleatorizados a uno de cuatro grupos de tratamiento: ezetimiba y simvastatina coadministradas equivalente a VYTORIN (10/10, 10/20, y 10/40), o simvastatina 20 mg. Los pacientes que no lograban un C-LDL < 100 mg/dL tuvieron su dosis de simvastatina titulada a intervalos de 6 semanas hasta una dosis máxima de 80 mg. En la Semana 5, las reducciones de C-LDL con VYTORIN 10/10, 10/20 o 10/40 fueron significativamente mayores que con simvastatina 20 mg. Además, en la Semana 5, un número significativamente mayor de pacientes que recibieron VYTORIN 10/10, 10/20, o 10/40 lograron la meta de C-LDL, en comparación con los que recibieron simvastatina 20 mg (ver Tabla 2). Los resultados de la Semana 5 para la reducción de C-LDL y el logro de la meta de C-LDL fueron consistentes con los resultados del final del estudio (Semana 23).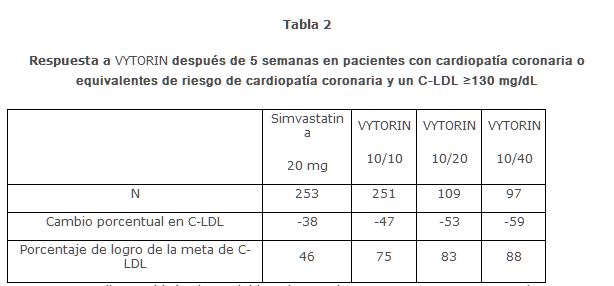
En un estudio multicéntrico, doble ciego, de 6 semanas, 1,902 pacientes con hipercolesterolemia primaria, que no habían cumplido con su meta de C-LDL objetivo del Programa Nacional de Educación sobre Colesterol (NCEP) ATP III, fueron aleatorizados a uno de ocho grupos de tratamiento: VYTORIN (10/10, 10/20, 10/40, o 10/80) o atorvastatina (10 mg, 20 mg, 40 mg u 80 mg). Cuando los pacientes que recibieron todas las dosis de VYTORIN fueron comparados con los que recibieron todas las dosis de atorvastarina, VYTORIN redujo significativamente el C-total, Apo B, y C-no-HDL y aumentó C-HDL significativamente más que atorvastatina. Los efectos de VYTORIN sobre los TG fueron similares a los efectos observados con atorvastatina. (Ver Tabla 3).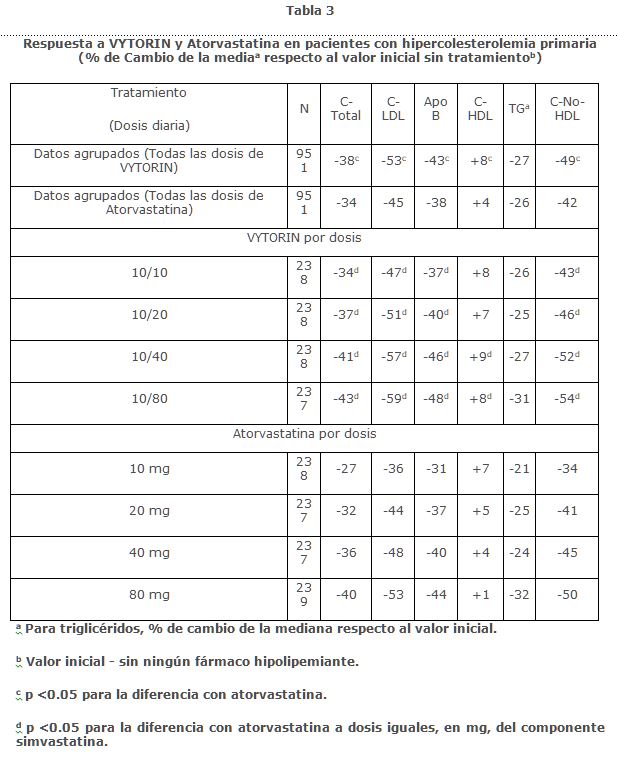
En un estudio multicéntrico, doble ciego, de 24 semanas, de ajuste forzado de la dosificación, en 788 pacientes con hipercolesterolemia primaria, que no habían cumplido con su meta de C-LDL objetivo del NCEP ATP III, fueron aleatorizados para recibir ezetimiba co-administrada con simvastatina equivalente a VYTORIN (10/10 y 10/20) o atorvastatina 10 mg. Para los tres grupos de tratamiento, la dosis de la estatina se tituló a intervalos de 6 semanas a 80 mg. En cada comparación de dosis pre-especificada, VYTORIN redujo el C-LDL en un mayor grado que atorvastatina (ver Tabla 4).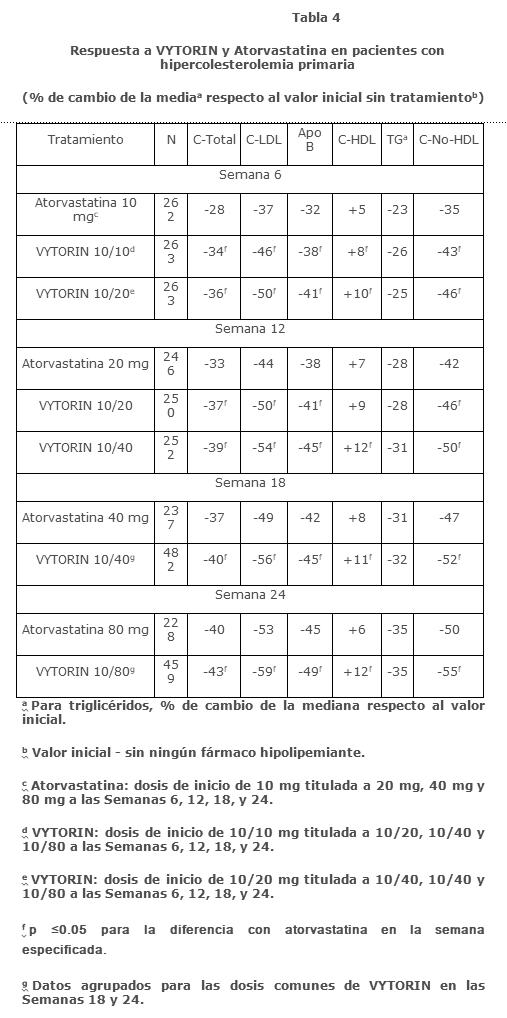
En un estudio multicéntrico, doble ciego, de 6 semanas, 2,959 pacientes con hipercolesterolemia primaria, que no habían cumplido con su meta de C-LDL objetivo del NCEP ATP III, fueron aleatorizados a uno de seis grupos de tratamiento: VYTORIN (10/20, 10/40, o 10/40 o 10/80) o rosuvastatina (10 mg, 20 mg o 40 mg). Cuando los pacientes que recibieron todas las dosis de VYTORIN fueron comparados con los que recibieron todas las dosis de rosuvastatina, VYTORIN redujo C-total, C-LDL, Apo B, TG y C-no-HDL, significativamente más que rosuvastatina. Los efectos de VYTORIN sobre el C- HDL fueron similares a los efectos observados con rosuvastatina. (Ver Tabla 5).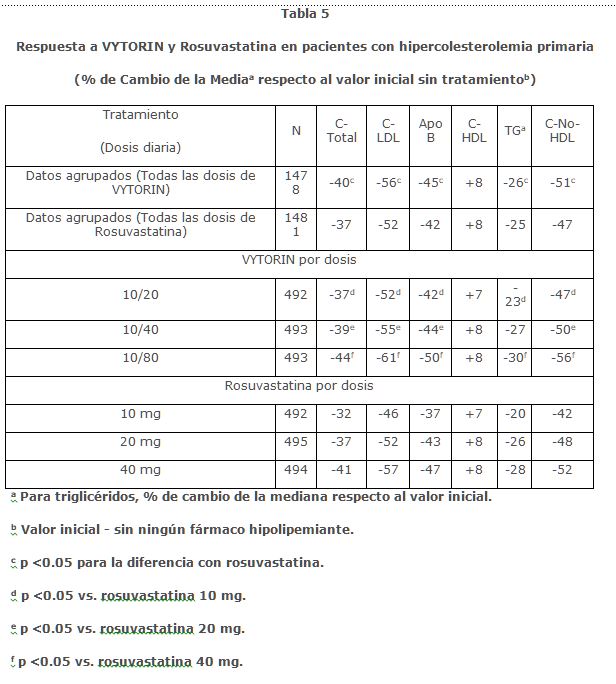
En un estudio doble ciego, controlado con placebo, de 8 semanas, 240 pacientes con hipercolesterolemia que ya recibían monoterapia con simvastatina y no cumplían con la meta de C-LDL del Programa Nacional de Educación sobre el Colesterol (NCEP) (2.6 a 4.1 mmol/L [100 a 160 mg/dL], dependiendo de las características basales) fueron aleatorizados para recibir ya sea ezetimiba 10 mg o placebo además de su terapia de simvastatina en curso. Entre los pacientes tratados con simvastatina que no cumplían la meta de C-LDL a nivel basal (~80%), significativamente más pacientes aleatorizados a la coadministración de ezetimiba con simvastatina alcanzaron su meta de C-LDL en el punto final del estudio en comparación con los pacientes aleatorizados a placebo coadministrado con simvastatina, 76% y 21.5%, respectivamente. Las reducciones correspondientes de C-LDL para ezetimiba o placebo coadministrado con simvastatina también fueron significativamente diferentes (27% o 3%, respectivamente). Además, ezetimiba coadministrada con simvastatina redujo significativamente el C-total, Apo B y TG en comparación con el placebo coadministrado con simvastatina. En un estudio multicéntrico, doble ciego, de 24 semanas, 214 pacientes con diabetes mellitus tipo 2 tratados con tiazolidinedionas (rosiglitazona o pioglitazona) durante un mínimo de 3 meses y simvastatina 20 mg durante un mínimo de 6 semanas, con un C-LDL promedio de 93 mg/dL, se aleatorizaron para recibir ya sea 40 mg de simvastatina o los ingredientes activos coadministrados equivalentes a VYTORIN 10/20. VYTORIN 10/20 fue significativamente más efectivo que duplicar la dosis de simvastatina de 40 mg en una mayor reducción de C-LDL (-21% y 0%, respectivamente), C-total (-14% y -1%, respectivamente), Apo B (-14% y -2%, respectivamente), y C-no-HDL (-20% y -2%, respectivamente) más allá de las reducciones observadas con simvastatina 20 mg. Los resultados para C-HDL y TG entre los dos grupos de tratamiento no fueron significativamente diferentes. Los resultados no se vieron afectados por el tipo de tratamiento de tiazolidinedionas. Coadministración con fenofibrato En un estudio clínico multicéntrico, doble ciego, controlado con placebo, en pacientes con hiperlipidemia mixta, 611 pacientes fueron tratados durante un máximo de 12 semanas. Los pacientes fueron aleatorizados para recibir placebo, VYTORIN 10/20 solo, fenofibrato 160 mg solo, o VYTORIN 10/20 y fenofibrato 160 mg. VYTORIN coadministrado con fenofibrato redujo significativamente el C-total, C-LDL, Apo B, C-no-HDL y TG, en comparación con fenofibrato administrado solo y redujo significativamente Apo B, C-no- HDL y TG, y aumentó C-HDL en comparación con VYTORIN administrado solo. (Ver Tabla 6).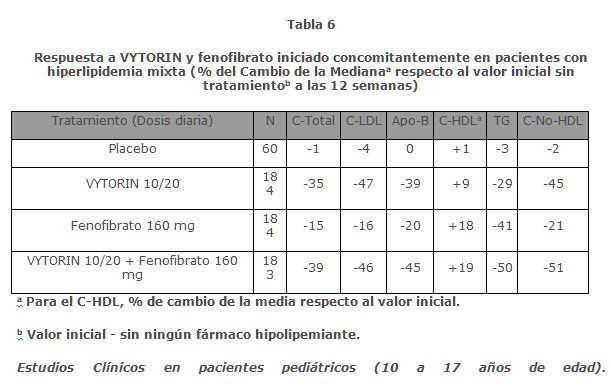
En un estudio multicéntrico, doble ciego, controlado, 142 niños y 106 niñas post-menárquicas, de 10 a 17 años de edad (edad promedio de 14.2 años) con hipercolesterolemia familiar heterocigótica (HeFH) fueron aleatorizados para recibir ya sea la coadministración de ezetimiba y simvastatina equivalente a VYTORIN o simvastatina sola. La inclusión en el estudio requirió 1) un nivel basal de C-LDL entre 160 y 400 mg/dL y 2) historia médica y presentación clínica compatible con HeFH. Los pacientes que recibieron VYTORIN (10/10, 10/20 o 10/40) o simvastatina (10 mg, 20 mg o 40 mg) durante 6 semanas, VYTORIN 10/40 o simvastatina 40 mg durante las siguientes 27 semanas y VYTORIN de manera abierta 10/10, 10/20 o 10/40 durante 20 semanas a partir de entonces. En la Semana 6, VYTORIN (todas las dosis) redujo el C-total, C-LDL, Apo B, y C-no-HDL significativamente más que simvastatina (todas las dosis). Resultados para TG y C-HDL fueron similares para los dos grupos de tratamiento. (Ver Tabla 7) En la Semana 33, VYTORIN redujo C-total, C-LDL, Apo B, TG y C-no-HDL significativamente más que simvastatina. Los aumentos de C-HDL fueron similares para los dos grupos de tratamiento. Además en la semana 33, un número significativamente mayor de pacientes que recibieron VYTORIN 10/40 (63%) alcanzaron la meta ideal de la Academia Americana de Pediatría (AAP) para C-LDL ( < 110 mg/dL) en comparación con los que recibieron simvastatina 40 mg (27%). En la Semana 53, los porcentajes promedio de cambio respecto al valor inicial para todas las dosis de VYTORIN fueron: -39% (C-total); -49% (C-LDL); -23% (Apo B); +3% (C-HDL); -17% (TG); y -46% (C-no-HDL).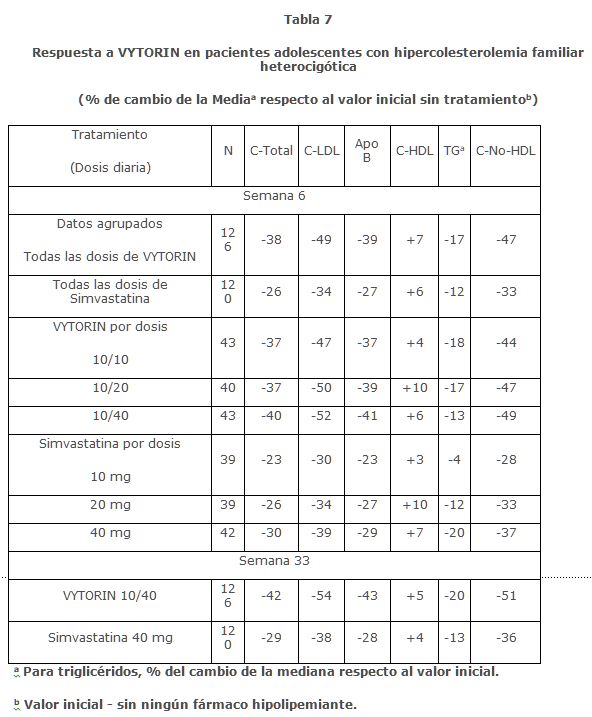
La seguridad y eficacia de las dosis por encima de 10/40 mg al día no se han estudiado en niños. La eficacia a largo plazo de la terapia con VYTORIN en la infancia, para reducir la morbilidad y la mortalidad en la edad adulta, no se ha estudiado. Ezetimiba En dos estudios, multicéntricos, doble ciego, controlados con placebo, de 12 semanas, en 1719 pacientes con hipercolesterolemia primaria, ezetimiba redujo significativamente C-total (13%), C-LDL (19%), Apo B (14%), y TG (8%) y aumentó C-HDL (3%) en comparación con placebo. La reducción de C-LDL era consistente entre edad, género, raza, y C-LDL basal. Además, ezetimiba no tuvo efecto sobre las concentraciones plasmáticas de las vitaminas A, D, y E liposolubles, no tuvo ningún efecto sobre el tiempo de protrombina, y no afectó negativamente la producción de hormona esteroide adrenocortical. Simvastatina VYTORIN contiene simvastatina. En dos grandes estudios clínicos controlados con placebo, el Scandinavian Simvastatin Survival Study (N = 4,444 pacientes) y el Heart Protection Study (N = 20,536 pacientes), los efectos del tratamiento con simvastatina fueron valorados en pacientes con alto riesgo de eventos coronarios, debido a enfermedades coronarias, diabetes, enfermedad vascular periférica, antecedentes de apoplejía, u otra enfermedad cerebrovascular existentes. La simvastatina demostró reducir: el riesgo de mortalidad total mediante la reducción de mortalidad por cardiopatía coronaria, el riesgo de infarto de miocardio no fatal y apoplejía, y la necesidad de procedimientos de revascularización coronaria y no coronaria. El beneficio adicional de VYTORIN en la morbilidad y mortalidad cardiovascular sobre y por encima de lo demostrado para simvastatina, no se ha establecido. Hipercolesterolemia familiar homocigótica (HoFH) Se realizó un estudio doble ciego, aleatorizado, de 12 semanas, en pacientes con diagnóstico clínico y/o genotípico de HoFH. Los datos se analizaron a partir de un subgrupo de pacientes (n = 14) que recibieron simvastatina 40 mg a nivel basal. El aumento de la dosis de simvastatina de 40 a 80 mg (n = 5) produjo una reducción de C-LDL de 13% respecto al valor inicial con simvastatina 40 mg. La coadministración de ezetimiba y simvastatina equivalente a VYTORIN (10/40 y 10/80 agrupados, n = 9), produjo una reducción de C-LDL de 23% respecto al valor inicial con simvastatina 40 mg. En aquellos pacientes con coadministración de ezetimiba y simvastatina equivalente a VYTORIN (10/80, n = 5), se produjo una reducción de C-LDL de 29% respecto al valor inicial con simvastatina 40 mg.
Contraindicaciones: Hipersensibilidad a cuaquiera de los componentes de este producto. Enfermedad hepática activa o aumentos persistentes inexplicables de las transaminasas séricas. Embarazo y lactancia (ver Restricciones de uso durante el embarazo y la lactancia). Cuando VYTORIN se va a administrar con fenofibrato, por favor consulte el la Información para Prescribir de fenofibrato. La administración concomitante de inhibidores potentes de CYP3A4 (p. ej., itraconazol, ketoconazol, posaconazol, voriconazol, inhibidores de la proteasa del VIH, boceprevir, telaprevir, eritromicina, claritromicina, telitromicina, nefazodona, y los fármacos que contienen cobicistat) (ver Precauciones generales, Miopatía / Rabdomiólisis y Interacciones medicamentosas y de otro género). Administración concomitante de gemfibrozilo, ciclosporina o danazol (ver Precauciones generales, Miopatía / Rabdomiólisis y Interacciones medicamentosas y de otro género).
Precauciones generales: Cuando VYTORIN se va a administrar con fenofibrato, por favor consulte la Información para Prescribir de fenofibrato. Miopatía / rabdomiólisis Simvastatina, como otros inhibidores de la HMG-CoA reductasa, causa ocasionalmente miopatía, manifestada como dolor muscular, hiperestesia o debilidad, con creatina cinasa (CK) por encima de 10 veces el límite superior normal (LSN). La miopatía algunas veces toma la forma de rabdomiólisis con o sin insuficiencia renal aguda secundaria a mioglobinuria, y en raras ocasiones se han producido muertes. El riesgo de miopatía aumenta con niveles altos de la actividad inhibitoria de la HMG-CoA reductasa en el plasma (p. ej., niveles elevados de simvastatina y ácido de simvastatina en plasma), lo cual puede deberse, en parte, a una interacción medicamentosa que interfiere con el metabolismo de simvastatina y/o con las vías de transportadores (ver Interacciones medicamentosas y de otro género). Los factores predisponentes a la miopatía incluyen edad avanzada (≥ 65 años), sexo femenino, hipotiroidismo no controlado, y la insuficiencia renal. Al igual que con otros inhibidores de la HMG-CoA reductasa, el riesgo de miopatía / rabdomiólisis está relacionado con la dosis para simvastatina. En una base de datos de estudios clínicos en los que 41,413 pacientes fueron tratados con simvastatina, 24,747 (aproximadamente el 60%) de los cuales fueron incluidos en estudios con una mediana de seguimiento de al menos 4 años, la incidencia de miopatía fue aproximadamente de 0.03%, 0.08% y 0.61% a los 20, 40 y 80 mg/día, respectivamente. En estos estudios, los pacientes fueron supervisados cuidadosamente y fueron excluidos algunos medicamentos interactuantes. En un estudio clínico en el que los pacientes con antecedentes de infarto de miocardio fueron tratados con simvastatina 80 mg/día (seguimiento promedio de 6.7 años), la incidencia de miopatía fue aproximadamente de 1.0% en comparación con 0.02% en los pacientes con 20 mg/día. Aproximadamente la mitad de estos casos de miopatía se produjeron durante el primer año de tratamiento. La incidencia de miopatía durante cada año de tratamiento posterior fue de aproximadamente 0.1%. El riesgo de miopatía es mayor en pacientes con simvastatina 80 mg en comparación con otras terapias basadas en estatinas con una eficacia similar de reducción de C-LDL. Por lo tanto, la dosis de 10/80 mg de VYTORIN sólo se debe utilizar en pacientes con alto riesgo de sufrir complicaciones cardiovasculares que no han alcanzado sus metas de tratamiento con dosis más bajas y cuando se espera que los beneficios superen a los riesgos potenciales. En pacientes que toman VYTORIN 10/80 mg para quienes necesitan un agente de interacción, se debe utilizar una dosis más baja de VYTORIN o un esquema alternativo de estatina-ezetimiba con menor potencial de interacciones farmacológicas (ver Contraindicaciones y Dosis y vía de adminstración). Todos los pacientes que inician tratamiento con VYTORIN, o cuya dosis de VYTORIN se está aumentando, deben ser advertidos del riesgo de miopatía y se les debe decir que reporten rápidamente cualquier dolor muscular inexplicable, hiperestesia o debilidad. La terapia con VYTORIN debe suspenderse inmediatamente si se diagnostica o se sospecha de miopatía. La presencia de estos síntomas, y un nivel de CK > 10 veces el límite superior normal, indican miopatía. En la mayoría de los casos, cuando los pacientes suspenden inmediatamente el tratamiento de simvastatina, los síntomas musculares y los aumentos de CK se resuelven (ver Reacciones secundarias y adversas). Se pueden considerar las determinaciones periódicas de CK en pacientes que inician tratamiento con VYTORIN o cuya dosis se aumenta. Se recomiendan determinaciones periódicas de CK para pacientes titulados a la dosis de 10/80 mg. No hay garantía de que esa supervisión prevenga la miopatía. Muchos de los pacientes que han desarrollado rabdomiólisis durante la terapia con simvastatina han tenido antecedentes médicos complicados, incluyendo insuficiencia renal por lo general como consecuencia de diabetes mellitus de larga evolución. Estos pacientes que toman VYTORIN requieren una supervisión más estrecha. La terapia con VYTORIN debe suspenderse temporalmente unos pocos días antes de una cirugía mayor programada y cuando surge cualquier condición médica o quirúrgica importante. En un estudio clínico en el que los pacientes con alto riesgo de enfermedad cardiovascular fueron tratados con simvastatina 40 mg/día (mediana de seguimiento de 3.9 años), la incidencia de miopatía fue aproximadamente de 0.05% para los pacientes no Chinos (n = 7,367) en comparación con 0.24% en los pacientes Chinos (n = 5,468). Aunque solo la población Asiática valorada en este estudio clínico era China, se debe tener precaución cuando se receta VYTORIN a pacientes Asiáticos y se debe emplear la menor dosis necesaria. Interacciones farmacológicas Debido a que VYTORIN contiene simvastatina, el riesgo de miopatía / rabdomiólisis aumenta por el uso concomitante de VYTORIN con los siguientes fármacos: Fármacos contraindicados. Inhibidores potentes de CYP3A4: Está contraindicado el uso concomitante con otros medicamentos con un potente efecto inhibidor sobre CYP3A4 en dosis terapéuticas (p. ej., itraconazol, ketoconazol, posaconazol, voriconazol, eritromicina, claritromicina, telitromicina, inhibidores de la proteasa del VIH, boceprevir, telaprevir, nefazodona, o fármacos que contienen cobicistat). Si el tratamiento a corto plazo con inhibidores potentes de CYP3A4 es inevitable, la terapia con VYTORIN debe suspenderse durante el curso del tratamiento. (Ver Farmacocinética y farmacodinamia, Propiedades Farmacocinéticas, Contraindicaciones, Interacciones medicamentosas y de otro género). Gemfibrozilo, ciclosporina o danazol: Está contraindicado el uso concomitante de estos fármacos con VYTORIN (Ver Farmacocinética y farmacodinamia, Propiedades Farmacocinéticas, Contraindicaciones, Interacciones medicamentosas y de otro género). Otros Fármacos Ácido fusídico: Los pacientes tratados con ácido fusídico y tratados concomitantemente con simvastatina pueden tener un mayor riesgo de Miopatía / Rabdomiólisis (ver Interacciones medicamentosas y de otro género, Otras interacciones farmacológicas). No se recomienda la coadministración con ácido fusídico. En pacientes en los que se considera esencial el uso de ácido fusídico sistémico, VYTORIN debe interrumpirse durante toda la duración del tratamiento con ácido fusídico. En circunstancias excepcionales, cuando se necesita ácido fusídico sistémico de manera prolongada, por ejemplo para el tratamiento de infecciones graves, la necesidad de la administración conjunta de VYTORIN y ácido fusídico sólo debe considerar caso por caso bajo estrecha supervisión médica. Amiodarona: En un estudio clínico, se reportó miopatía en el 6% de los pacientes que recibían simvastatina 80 mg y amiodarona. La dosis de VYTORIN no debe superar los 10/20 mg al día en pacientes que reciben medicamentos con amiodarona. (Ver Interacciones medicamentosas y de otro género). Bloqueadores de canales de calcio Verapamilo o diltiazem: Los pacientes que toman diltiazem tratados concomitantemente con 80 mg de simvastatina tuvieron un mayor riesgo de miopatía. La dosis de VYTORIN no debe superar los 10/20 mg al día en pacientes que reciben medicamentos concomitantes con verapamilo o diltiazem. (Ver Interacciones medicamentosas y de otro género, Otras interacciones farmacológicas.) Amlodipino: En un estudio clínico, pacientes con amlodipino tratados concomitantemente con 80 mg de simvastatina tuvieron un ligero incremento del riesgo de miopatía (ver Interacciones medicamentosas y de otro género). La dosis de VYTORIN no debe superar los 10/40 mg al día en pacientes que reciben medicamentos con amlodipino. Lomitapida: La dosis de VYTORIN no debe superar los 10/40 mg al día en pacientes con HoFH que reciben medicamentos concomitantes con lomitapida (ver Interacciones medicamentosas y de otro género). Inhibidores moderados de CYP3A4: Los pacientes que toman otros medicamentos que declaran tener un efecto inhibidor moderado sobre CYP3A4 concomitantemente con VYTORIN, particularmente las dosis más altas de VYTORIN pueden tener un mayor riesgo de miopatía. Cuando se coadministra VYTORIN con un inhibidor moderado de CYP3A4, puede ser necesario un ajuste de la dosis de VYTORIN. Inhibidores de la Proteína de Resistencia a Cáncer de Mama (BCRP, por las siglas en inglés para Breast Cancer Resistant Protein): La administración concomitante de productos que son inhibidores de BCRP (p.ej., elbasvir y grazoprevir) pueden ocasionar incremento en las concentraciones plasmáticas de simvastatina y un incremento en el riesgo de miopatía; por lo tanto, puede ser necesario un ajuste de la dosis de VYTORIN. No se ha estudiado la co-administración de elbasvir y grazoprevir con simvastatina; sin embargo, la dosis de VYTORIN no debe exceder de 10/20 mg diarios en pacientes que reciben medicación concomitante con productos que contienen elbasvir o grazoprevir (ver Interacciones medicamentosas y de otro género, Otras interacciones farmacológicas). Fenofibrato: En un estudio en el que VYTORIN 10/20 mg/día y fenofibrato 160 mg/día se coadministraron en 183 pacientes hasta por 12 semanas, no hubo reportes de miopatía. No se han estudiado dosis de VYTORIN por arriba de 10/20 mg/día y fenofibrato. Se debe tener precaución cuando se prescriben VYTORIN y fenofibrato, ya que fenofibrato puede provocar miopatía cuando se administra solo. En otro estudio de 12 semanas, en el que 411 pacientes recibieron simvastatina 20 mg/día y fenofibrato 160 mg/día, la coadministración fue también bien tolerada. Si se sospecha de colelitiasis en un paciente que recibe VYTORIN y fenofibrato, se indican estudios de la vesícula biliar y se debe considerar una terapia hipolipemiante alternativa (ver Reacciones secundarias y adversas y la Información para Prescribir de fenofibrato). Otros Fibratos: No se han estudiado la seguridad y efectividad de VYTORIN administrado con fibratos, excepto fenofibrato. Por lo tanto, el uso concomitante de VYTORIN y fibratos, excepto fenofibrato, debe evitarse. Está contraindicado el uso concomitante de gemfibrozilo (ver Contraindicaciones). Niacina (≥1 g/día): Se han observado casos de miopatía/rabdomiólisis con simvastatina coadministrada con niacina en dosis modificadoras de lípidos (≥ 1g/día). En un estudio clínico (mediana de seguimiento de 3.9 años) con pacientes con alto riesgo de enfermedad cardiovascular y con los niveles de C-LDL bien controlados con simvastatina 40 mg/día, con o sin ezetimiba 10 mg, no hubo beneficio adicional en los desenlaces cardiovasculares con la adición de dosis modificadoras de lípidos (≥1 g/día) de niacina. Por lo tanto, el beneficio del uso combinado de simvastatina con niacina debe ser sopesado cuidadosamente con los riesgos potenciales de la combinación. Además, en este estudio, la incidencia de miopatía fue de aproximadamente 0.24% para los pacientes Chinos con simvastatina 40 mg o ezetimiba/simvastatina 10/40 mg en comparación con el 1.24% para los pacientes chinos con simvastatina 40 mg o ezetimiba/simvastatina 10/40 mg coadministrados con niacina/laropiprant de liberación prolongada 2 g/40 mg. Mientras que la única población Asiática valorada en este estudio clínico era China, debido a que la incidencia de miopatía es mayor en los pacientes Chinos que en los no Chinos, no se recomienda la coadministración con VYTORIN con dosis modificadoras de lípidos (≥1 g/día) de niacina en pacientes Asiáticos. (Ver Interacciones medicamentosas y de otro género). Daptomicina: Se han observado reportes de miopatía y/o rabdomiólisis con inhibidores de HMG-CoA reductasa coadministrados con daptomicina. Se debe tener precaución al prescribir inhibidores de HMG- CoA reductasa con daptomicina, porque cualquiera de los dos agentes puede causar miopatía y/o rabdomiólisis cuando se administra solo. Se debe considerar suspender VYTORIN de forma temporal en pacientes que toman daptomicina (ver Interacciones medicamentosas y de otro género). Anticoagulantes: Si VYTORIN se añade a warfarina, a otro anticoagulante cumarínico, o fluindiona, se debe supervisar adecuadamente la Relación Normalizada Internacional (INR) (ver Interacciones medicamentosas y de otro género). Enzimas hepáticasEn los estudios de coadministración controlados en pacientes que recibieron ezetimiba con simvastatina, se observaron elevaciones consecutivas de las transaminasas (≥3 X LSN). (Ver Reacciones secundarias y adversas). Se recomienda que se realicen pruebas del funcionamiento hepático antes del tratamiento con VYTORIN y después cuando esté clínicamente indicado. Los pacientes titulados a la dosis de 10/80 mg deben realizarse una prueba adicional antes de la titulación, 3 meses después de la titulación a la dosis de 10/80 mg, y posteriormente de forma periódica (por ejemplo, cada seis meses) durante el primer año de tratamiento. Se debe prestar especial atención a los pacientes que desarrollan niveles elevados de transaminasas séricas, y en estos pacientes, las mediciones se deben repetir pronto y con mayor frecuencia. Si los niveles de transaminasas muestran evidencia de progresión, especialmente si se elevan a 3 veces LSN y son persistentes, el fármaco debe ser descontinuado. Tenga en cuenta que ALT puede emanar de los músculos, por lo tanto, el aumento de ALT con CK puede indicar miopatía (ver Precauciones generales, Miopatía/Rabdomiólisis). Ha habido raros reportes posteriores a la comercialización de insuficiencia hepática fatal y no fatal en pacientes que toman estatinas, incluyendo simvastatina. Si ocurre lesión hepática grave con síntomas clínicos y/o hiperbilirrubinemia o ictericia durante el tratamiento con VYTORIN, interrumpir la terapia con prontitud. Si no se encuentra una etiología alternativa, no reinicie VYTORIN. VYTORIN debe utilizarse con precaución en pacientes que consumen cantidades importantes de alcohol y/o tienen antecedentes de enfermedad hepática. Las enfermedades hepáticas activas o los incrementos inexplicables persistentes de las transaminasas son contraindicaciones para el uso de VYTORIN. Insuficiencia hepática Debido a los efectos desconocidos de una mayor exposición a ezetimiba en pacientes con insuficiencia hepática moderada o grave, no se recomienda VYTORIN en estos pacientes (ver Farmacocinética y farmacodinamia - Características en grupos de pacientes especiales). Uso Pediátrico La seguridad y efectividad de VYTORIN en los pacientes de 10 a 17 años de edad con hipercolesterolemia familiar heterocigótica se han evaluado en un estudio clínico controlado en niños y en las niñas adolescentes que tenían al menos un año después de la menarquia. Los pacientes adolescentes tratados con VYTORIN tenían un perfil similar de experiencias adversas a la de los pacientes adultos tratados con VYTORIN. Las dosis superiores a 10/40 mg/día no se han estudiado en esta población. En este estudio controlado, no hubo ningún efecto detectable sobre el crecimiento o la maduración sexual en los niños o niñas adolescentes, o ningún efecto sobre la duración del ciclo menstrual en niñas. (Ver V. Estudios Clínicos, Estudios Clínicos en Pacientes Pediátricos (10 a 17 años de edad); Reacciones secundarias y adversas y Dosis y vía de adminstración). VYTORIN no ha sido estudiado en pacientes menores de 10 años de edad o en niñas pre-menárquicas. Uso en Pacientes de Edad Avanzada Debido a que la edad avanzada (≥65 años) es un factor de predisposición a la miopatía, VYTORIN debe prescribirse con precaución en los adultos de edad avanzada. En un estudio clínico de los pacientes tratados con simvastatina 80 mg/día, los pacientes ≥65 años de edad tuvieron un mayor riesgo de miopatía en comparación con pacientes menores de < 65 años de edad.
Restricciones de uso durante el embarazo y la lactancia: EMBARAZO La aterosclerosis es un proceso crónico, y normalmente la suspensión de los fármacos hipolipemiantes durante el embarazo debería tener poco impacto en el riesgo a largo plazo asociados con hipercolesterolemia primaria. VYTORIN VYTORIN está contraindicado durante el embarazo (ver Contraindicaciones, y Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).Simvastatina La seguridad de simvastatina en mujeres embarazadas no se ha establecido. No se han realizado estudios clínicos controlados con simvastatina en mujeres embarazadas. Rara vez se han recibido reportes de anomalías congénitas tras la exposición intrauterina a los inhibidores de la HMG-CoA reductasa. Sin embargo, en un análisis de aproximadamente 200 embarazos supervisados prospectivamente expuestos durante el primer trimestre a simvastatina o a otro inhibidor de la HMG-CoA reductasa estrechamente relacionado, la incidencia de anomalías congénitas fue comparable a la observada en la población general. Este número de embarazos fue estadísticamente suficiente para excluir un aumento de 2.5 veces o más en las anomalías congénitas sobre la incidencia general. Aunque no hay evidencia de que la incidencia de anomalías congénitas en los descendientes de los pacientes que toman simvastatina u otro inhibidor de la HMG-CoA reductasa estrechamente relacionado sea diferente de la observada en la población general, el tratamiento materno con simvastatina puede reducir los niveles fetales de meval