WILATE
OCTAPHARMA
Denominación genérica: Factor VIII (Factor Antihemofílico A), Factor von Willebrand.
Forma farmacéutica y formulación: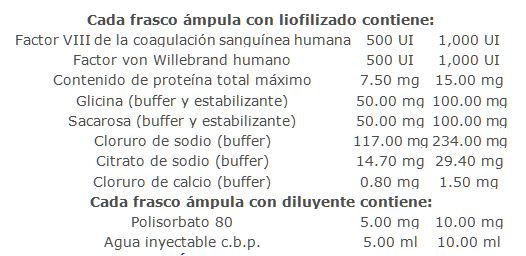
Indicaciones terapéuticas: Enfermedad Von Willebrand: Tratamiento y profilaxis de sangrado en pacientes con enfermedad Von Willebrand (EvW) debido a la deficiencia cualitativa o cuantitativa en FvW, cuando el tratamiento con desmopresina (1-deamino-8-D-Arginina vasopresina [DDAVP]) es ineficaz o está contraindicado.Indicaciones mayores: Prevención y tratamiento de episodios de sangrado, y Prevención y tratamiento de sangrado en cirugía menor.Hemofilia A: Tratamiento y profilaxis de sangrado en pacientes con hemofilia A (deficiencia congénita de FVIII) y para la prevención y tratamiento de sangrado en procedimientos de cirugías menores.Los ensayos clínicos controlados para evaluar la seguridad y eficacia de WILATE en cirugías mayores actualmente continúan en pacientes con enfermedad de Von Willebrand y en pacientes con hemofilia tipo A. Por consiguiente, los datos disponibles están limitados actualmente, los cuales evalúan o revisan las recomendaciones de dosificación. Así en el caso de intervenciones quirúrgicas mayores, es indispensable un preciso monitoreo de la sustitución de terapia por medio del análisis de la coagulación (FVIII-C y posiblemente FvW:RCo).
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: Enfermedad de von Willebrand: El factor de Von Willebrand (FvW) en un concentrado, es un constituyente normal del plasma humano y se comporta de la misma manera que un FvW endógeno.La administración de FvW permite la corrección de las anormalidades hemostásicas presentadas en pacientes que padecen la deficiencia de factor de Von Willebrand en dos niveles: El FvW reestablece la adhesión plaquetaria al sub-endotelio vascular en el sitio de daño vascular (como en la unión de ambos al sub-endotelio vascular y en la membrana plaquetaria) proporcionando en la hemostasia primaria la reducción del tiempo de sangrado. Este efecto ocurre inmediatamente y es conocido para depender a una gran extensión de la polimerización de la proteína. El FvW produce una corrección prolongada de la asociación de la deficiencia de FVIII. Administrada intravenosamente, el FvW liga, el FVIII endógeno (el cual es normalmente producido por el paciente) y estabiliza este factor, evitando su degradación rápida.Debido a esto, la administración pura de FvW (concentrado de FvW con bajos niveles de FVIII) restaura el FVIII: Nivel C a normal como un efecto secundario después de la primera infusión.El FvW, facilita la agregación plaquetaria por medio de la unión al receptor plaquetario glicoproteína IIb/IIIa.La administración del concentrado de FVIII/FvW restaura el FVIII a niveles normales inmediatamente después de la primera infusión.En resumen el FvW funciona como una proteína de protección al FVIII, media la adhesión plaquetaria a sitios de daño vascular y facilita la agregación plaquetaria.Hemofilia A: El complejo FVIII/FvW consiste en dos moléculas (FVIII y FvW) con diferentes funciones fisiológicas diferentes. Cuando es infundido a un paciente con hemofilia el FVIII se une al FvW en la circulación del paciente. El FVIII activado (FVIIIa) actúa como un cofactor para activar al FX (FIXa), acelerando la conversión a factor X para activar el factor X (FXa). FXa convierte la protrombina en trombina. La trombina entonces convierte el fibrinógeno en fibrina y entonces se forma el coágulo. La hemofilia A es un desorden de la coagulación sanguínea hereditario ligado al sexo, debido a la disminución en los niveles de FVIII, resultando sangrados profusos en las articulaciones, músculos u órganos internos, o sangrados espontáneos como resultado de accidentes o traumas quirúrgicos. Con la terapia de reemplazo del plasma se incrementan los niveles de FVIII, de manera temporal de la deficiencia de factor y corrección de la tendencia de sangrado.Propiedades farmacocinéticas:Enfermedad de von Willebrand: En la enfermedad de Von Willebrand. En pacientes con EvW tipo 3, para FvW:RCo y FvW:Ag tiene un valor de recuperación media de 68 y 99% siendo calculados respectivamente. Estos valores corresponden a los incrementos medios en plasma de 1.5 y 2.1% por UI/kg de peso sustituido.Los resultados siguientes fueron observados en un estudio clínico que involucra 8 pacientes con EvW tipo 3: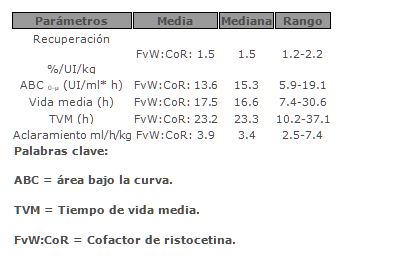
Hemofilia A: El FVIII (de un concentrado) es un constituyente normal del plasma humano y actúa como el FVIII endógeno.Después de la inyección del producto, aproximadamente de dos terceras a tres cuartas partes del FVIII permanece en la circulación. El nivel FVIII:C (procoagulante del FVIII) alcanzado en el plasma ha mostrado ser entre 80-120% en el predicto FVIII:C.FVIII:C: FVIII:C disminuye en dos fases su caída exponencial. En la fase inicial, la distribución entre los compartimientos intravasculares y otros compartimientos (líquidos corporales) ocurre con un tiempo de vida media de eliminación en plasma de 3 a 6 horas. En la fase subsecuente, fase lenta, la vida media varía entre 8 a 18 horas, con un promedio de 15 horas. Esto corresponde al tiempo de vida media real biológica.Los siguientes resultados fueron observados en un estudio clínico con 12 pacientes (ensayo de coagulación fase I, sólo las medidas).
Contraindicaciones: Hipersensibilidad a la sustancia activa, o a alguno de los excipientes.
Precauciones generales: Como cualquier infusión intravenosa de los productos derivados de las proteínas del plasma, las reacciones de hipersensibilidad del tipo alérgico se pueden presentar.Los pacientes deberán monitorearse cuidadosamente y estrechamente para observar cualquier aparición de síntomas durante el periodo de Infusión.Los pacientes deberán informar cualquier signo temprano de reacciones de hipersensibilidad como ronchas, urticaria generalizada, opresión torácica, hipotensión y anafilaxis.Si ocurrieran los síntomas alérgicos, se deberá descontinuar su administración inmediatamente y avisar al médico.En caso de shock, las medidas estándares médicas para tratamiento de shock deberán de llevarse a cabo.Las medidas estándares para prevenir la transmisión de infecciones del uso de medicamentos preparados de sangre y/o plasma humano, incluye la selección de donadores, pruebas a las donaciones individuales de marcadores específicos de infección y la inclusión efectiva de pasos en los procesos de manufactura para la inactivación/remoción de agentes virales. A pesar de esto, cuando los medicamentos preparados de sangre y/o plasma humano son administrados, la posibilidad de transmitir agentes infecciosos no puede quedar totalmente excluida. Esto también aplica a virus emergentes desconocidos y otros patógenos.Las medidas tomadas son consideradas efectivas para virus envueltos como VIH, VHB, y VHC, y para los virus no envueltos como el VHA, estas son limitadas contra los virus no envueltos como el parvovirus B19.La infección por parvovirus B19 puede ser muy seria en mujeres embarazadas (infección fetal) y para individuos con inmunodeficiencia o incremento en la eritropoyesis (por ejemplo, anemia hemolítica).Se recomienda ampliamente que cada vez que sea administrado WILATE a un paciente, el nombre y número de lote del producto se anote para mantener la rastreabilidad entre el paciente y el lote del producto.La vacunación apropiada (hepatitis A y B) podría ser considerada para pacientes que han recibido repetidamente o regularmente concentrados de FVIII/FvW derivados del plasma humano.De acuerdo con protocolos para el tratamiento de la enfermedad Von Willebrand, se deberán usar con precaución los agentes antiinflamatorios no esteroideos, y se deberá evitar la administración de medicamentos que provoquen disfunción plaquetaria, especialmente aquellos que contienen ácido acetilsalicílico (Aspirina®).Enfermedad de von Willebrand: Cuando se utilizan productos que contienen FVIII/FvW, el médico tratante debe de estar consciente que el tratamiento continuo puede causar un aumento excesivo en FVIII:C, en pacientes que reciben productos de FVIII que contienen FvW, los niveles de plasma de FVIII:C deben de monitorearse para evitar los niveles en plasma sostenidos de FVIII:C, los cuales podrían incrementar el riesgo de eventos tromboembólicos.Existe un riesgo de eventos trombóticos cuando se usan productos de FVIII que contienen FvW, particularmente en pacientes con factores de riesgo conocido de laboratorio y por clínica. Por consiguiente, deben monitorearse los pacientes con riesgo con los signos tempranos de trombosis. La profilaxis para el tromboembolismo venoso deberá instituirse de acuerdo a las recomendaciones actuales.Los pacientes con EvW, especialmente del tipo 3, pueden desarrollar anticuerpos neutralizantes (inhibidores) al FvW. Si los niveles esperados en plasma de la actividad de FvW:CoR no se logran, o si el sangrado no es controlado con las dosis apropiadas, deberá ser realizada una prueba de laboratorio para determinar si un inhibidor de FvW está presente. En pacientes con niveles altos de inhibidores, la terapia con FvW no podrá ser efectiva y otras opciones terapéuticas deberán ser consideradas.El manejo de tales pacientes deberá ser por médicos con experiencia en cuidado de desórdenes hemostásicos.Hemofilia A: La formación de anticuerpos neutralizantes (inhibidores) al FVIII es conocida como una complicación en el manejo de individuos con hemofilia A. Estos inhibidores son usualmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del FVIII, la cual es cuantificada en Unidades Bethesda (UB) por ml que utiliza el ensayo modificado. El riesgo de desarrollar inhibidores está correlacionado a la exposición del FVIII antihemofílico, este riesgo es más alto dentro de los primeros 20 días de exposición. Raramente, los inhibidores se pueden desarrollar después de los primeros 100 días de exposición.Los pacientes tratados con FVIII deberán estar cuidadosamente monitoreados por el desarrollo de inhibidores con observaciones clínicas apropiadas y pruebas de laboratorio.
Restricciones de uso durante el embarazo y la lactancia: No se han realizado estudios con animales en reproducción con FVIII/FvW, valorando riesgo-beneficio.Enfermedad de von Willebrand: La experiencia en el tratamiento con mujeres embarazadas o mujeres en periodo de lactancia no está disponible.WILATE deberá ser administrado a mujeres embarazadas o en periodo de lactancia con deficiencia de FvW sólo si está claramente indicado, tomando en consideración que la administración confiere un riesgo aumentado de eventos hemorrágicos en estos pacientes.Hemofilia A: Basados en la rara incidencia de hemofilia A en mujeres, la experiencia respecto al tratamiento durante el embarazo y el periodo de lactancia no está disponible. Por consiguiente, WILATE deberá usarse sólo si está claramente indicado.
Reacciones secundarias y adversas: Hipersensibilidad o reacciones alérgicas (la cual puede incluir angioedema, sensaciones de quemadura y picazón en el sitio de inyección, escalofríos, rubor, urticaria generalizada, cefalea, ronchas, hipotensión, letargo, náuseas, inquietud, taquicardia, opresión torácica, sensación de picazón, vómito y disnea) han sido observados muy raramente, y puede en algunos casos progresar a anafilaxis severa (incluyendo shock).En raras ocasiones, se ha observado fiebre.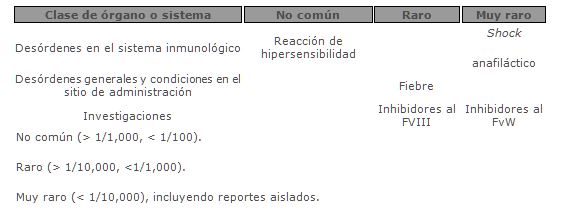
Enfermedad de von Willebrand: Los pacientes con EvW, especialmente la tipo 3, puede muy raramente desarrollar anticuerpos neutralizantes al FvW. Si esto ocurriera, la condición podría manifestarse como una respuesta clínica inadecuada. Tales anticuerpos podrían precipitar y puede ocurrir concomitantemente una reacción anafiláctica. Por consiguiente, deben evaluarse pacientes que experimenten reacciones anafilácticas por la presencia de un inhibidor.En todos estos casos, es recomendable que se contacte a un especialista en hemofilia. Existe un riesgo de que ocurran eventos trombóticos, particularmente en pacientes con factores de riesgo clínicos y por laboratorio.Por consiguiente, los pacientes con riesgo, deberán ser monitoreados para detectar signos tempranos de trombosis. La profilaxis contra tromboembolia venosa deberá ser instituida de acuerdo a la recomendaciones clínicas actuales.En pacientes que recibieron productos de FVIII que contienen FvW, los niveles sostenidos excesivamente de FVIII:C pueden incrementar el riesgo de eventos trombóticos.Hemofilia A: Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) al FVIII. Si estos inhibidores ocurrieran, la condición podría manifestarse como una respuesta clínica insuficiente. En ambos casos, se recomienda que un centro especializado en hemofilia sea contactado.Debido al número pequeño de pacientes tratados con WILATE, un reporte final en el desarrollo de inhibidores en pacientes tratados previamente no se ha podido realizar.Los datos para la incidencia de inhibidores en pacientes no tratados previamente no están disponibles.Existen datos insuficientes para recomendar el uso de WILATE en pacientes no tratados previamente.
Interacciones medicamentosas y de otro género: WILATE no deberá ser mezclado con otros medicamentos durante su administración.
Alteraciones en los resultados de pruebas de laboratorio: No se tienen datos que indiquen que WILATE tiene algún impacto en otros parámetros de laboratorio.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: El FVIII y FvW en WILATE son constituyentes normales del plasma humano y actúa como el FVIII/FvW endógeno.
Para la toxicidad, pruebas con dosis repetidas en animales no son factibles debido a la interferencia con el desarrollo de anticuerpos a proteínas humanas heterólogas.La experiencia clínica no proporciona información indirecta para efectos, tumorigénicos y mutagénicos para el humano por FVIII/FvW.WILATE contiene trazas de cantidades del producto químico tri(n-butil)fosfato y Octaxynol (Tritón X-100), el cual se usa para el tratamiento con S/D para la inactivación viral durante el proceso de manufactura. Sin embargo, los datos preclínicos revelaron que no existe riesgo especial en humanos, basados en estudios convencionales de dosis repetidas de toxicidad, genotoxicidad y desarrollo embrión-fetal.
Dosis y vía de administración: Vía de administración intravenosa.El tratamiento debe iniciarse bajo la supervisión de un médico experimentado en el tratamiento de desórdenes de la coagulación. La cantidad de WILATE (UI) para usarse por kg de peso (kg) deberá de calcularse con base en la potencia del FVIII de la etiqueta. El producto es solo para uso individual y el contenido total del frasco deberá ser administrado. En caso de no administrar todo el medicamento, deberá desecharse el sobrante. Enfermedad de von Willebrand: El radio entre el FVIII:C y el FvW:CoR es apróximadamente de 1:1, generalmente 1 UI/kg de peso FVIII:C y FvW:CoR aumenta su nivel en plasma de 1.5 a 2% de actividad normal para la proteína respectiva.Usualmente, cerca de 20 a 50 UI de WILATE/kg de peso es necesaria para lograr la hemostasia primaria.Esto elevará el FVIII:C y el FvW:CoR en los pacientes en apróximadamente 30 a 100%.Enfermedad de von Willebrand: Una dosis inicial de 50 a 80 UI de WILATE/kg de peso podrá ser requerida, especialmente en pacientes con EvW tipo 3, en donde el mantenimiento adecuado de sus niveles en plasma podría requerir mayores dosis que los otros tipos de EvW. Prevención de hemorragias en caso de cirugía o trauma severo: Para prevenir el sangrado en caso de cirugía, la administración de WILATE deberá iniciarse 30 minutos antes de procedimiento quirúrgico. En caso de cirugía electiva, el tratamiento podría iniciar de 12-24 horas antes de la cirugía y deberá repetirse 1 hora antes del procedimiento. Los niveles de FvW:CoR ³ 60 UI/dl (≥ 60%) y niveles de FVIII:C ≥ 40 UI/dl (≥ 40%) deben lograrse.Una dosis apropiada deberá ser readministrada cada 12-24 horas de tratamiento. La dosis y duración del tratamiento dependerá del estado clínico del paciente, el tipo y la severidad del sangrado y de ambos niveles de FVIII:C y FvW:CoR. En pacientes que recibieron productos de FVIII con FvW, los niveles de plasma de FVIII:C deberán ser monitoreados para revelar los niveles de plasma de FVIII:C excesivamente sostenidos, los cuales pueden incrementar el riesgo de eventos trombóticos, particularmente en pacientes con factores de riesgo conocidos por clínica o por laboratorio.En caso de que se observen niveles excesivos de FVIII:C, la reducción de la dosis y/prolongación del intervalo de dosis o el uso de productos que contengan FvW que contengan bajos niveles de FVIII deberán ser considerados. Hemofilia A: La dosificación y la duración de la terapia está basada en la cantidad de la deficiencia del factor, severidad y localización de la hemorragia, y del curso clínico de la enfermedad.El número de unidades administradas de FVIII está expresada en UI (Unidades Internacionales), las cuales están relacionadas a los estándares de la FMH de los productos de FVIII. La actividad en plasma se expresa como un porcentaje (relativo al plasma humano normal) o en UI (relativo a los estándares internacionales para el FVIII derivado del plasma). Una Unidad Internacional (UI) de actividad del FVIII es equivalente a la cantidad de FVIII en 1 ml de plasma humano normal.El cálculo para la dosis requerida se basa en que 1 UI de FVIII/kg de peso incrementa la actividad de FVIII plasmática de 1.5 a 2% del normal. Para calcular la dosis se requiere determinar la actividad del factor VIII en plasma y calcular la cantidad que se necesita incrementar. La dosis requerida se calcula por la siguiente fórmula: Unidades requeridas = Peso corporal (kg) x incremento del factor VIII requerido % (UI/dl) x 0.5 UI/kg: La cantidad administrada y la frecuencia de la aplicación siempre deberá estar orientada a la efectividad clínica de cada paciente, por caso individual. La siguiente tabla puede ser usada como guía de dosificación en casos de eventos hemorrágicos y quirúrgicos en pacientes adultos y niños mayores de 6 años.En el caso de los siguientes eventos hemorrágicos, el FVIII:C no deberá disminuir por debajo de los niveles de plasma correspondientes.
Durante el tratamiento deberá realizarse, la determinación apropiada de niveles FVIII:C y su manejo de acuerdo a las guías de dosificación, infusión y frecuencia de la repetición de las infusiones.En el caso de intervenciones quirúrgicas mayores en particular, un monitoreo preciso de la terapia de sustitución por medio de análisis de la coagulación (FVIII:C) es indispensable. Los pacientes pueden variar en su respuesta de manera individual al tratamiento con FVIII, mientras se logran los diferentes niveles en vivo de la recuperación y de las diferentes vidas medias demostradas. Profilaxis: Para la profilaxis de largo plazo contra hemorragias en los pacientes con Hemofilia A severa, dosis de 20-40 UI de WILATE/kg de peso deberán administrarse en intervalos de 2-3 días. En algunos casos, especialmente en pacientes jóvenes, dosis con intervalos cortos o con intervalos largos pueden ser necesarias.Hemofilia A: Los pacientes deberán ser monitoreados para el desarrollo de anticuerpos neutralizantes (inhibidores). Si la actividad del FVIII de los niveles del plasma no se ha logrado, o si el sangrado no se ha controlado con las dosis apropiadas, deberá realizarse un ensayo para determinar si está presente un inhibidor al FVIII. En pacientes con altas dosis de inhibidores, la terapia con FVIII podría no ser efectiva y otras opciones terapéuticas deben ser consideradas. El manejo de cada paciente deberá ser directamente por médicos con experiencia en el cuidado de pacientes con desórdenes de la hemostasia.La experiencia de tratamiento con WILATE en niños menores de 6 años de edad está limitada. Método de administración: Instrucciones para la reconstitución:1. Calentar el frasco ámpula con liofilizado y el frasco ámpula con diluyente con los frascos cerrados a temperatura ambiente. Esta temperatura deberá ser mantenida durante la reconstitución. Si se usa baño de agua para el calentamiento debe tenerse cuidado de que el agua no entre en contacto con los tapones de hule o las retapas de los frascos. La temperatura del baño de agua no debe de exceder de +37°C.2. Retire las retapas del frasco con liofilizado y del frasco con diluyente, y limpie los tapones de hule con los apósitos con alcohol.3. Retirar la cubierta protectora de la punta corta de la aguja de doble punta, asegurándose de no tocar la punta expuesta de la aguja. Entonces perfore el centro del tapón de goma del frasco con solvente con la aguja sostenida verticalmente. Para retirar el líquido total del frasco con diluyente, la aguja debe ser introducida dentro del tapón de hule en tal forma que apenas penetre el tapón y sea visible en el frasco.4. Quitar la cubierta protectora de la punta larga de la aguja, asegurandose de no tocar la punta expuesta de la aguja. Voltear el frasco con el diluyente encima del frasco con liofilizado y rápidamente perforar el centro de este frasco con la aguja larga. El vacío dentro del frasco del concentrado atrae el diluyente.5. Retirar la aguja de doble punta con el frasco del diluyente vacío del frasco con el liofilizado, rotar lentamente el frasco hasta que el liofilizado se disuelva completamente. WILATE se disuelve rápidamente a temperatura ambiente en una solución clara.La solución es clara o ligeramente opalescente. Si el concentrado no puede disolverse completamente, o se forman agregados, la preparación no deberá usarse. Instrucciones para su administración: Como medida de precaución, se debe tomar el pulso antes y durante la administración de factor VIII, si hubiera un aumento significativo del pulso, la velocidad de administración debe reducirse o interrumpirse. 1.Después de que el liofilizado ha sido reconstituido de la manera anteriormente descrita, retire la cubierta protectora de la aguja filtrante y perfore el tapón de goma del frasco del concentrado. 2. Retire la cubierta de la aguja filtrante y sujete la jeringa.3. Dé vuelta al frasco con la jeringa unida al revés y jale la solución dentro de la jeringa. 4. Limpie el lugar seleccionado para inyección con un apósito con alcohol. 5. Quite la aguja filtrante de la jeringa y conecte la aguja de la infusión de mariposa a la jeringa. 6. Inyecte la solución intravenosamente a una velocidad baja de infusión de 2-3 ml/min. Cualquier producto sin usar o material desechado deberá de disponerse de acuerdo con los requerimientos locales.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existen síntomas reportados de sobredosificación con FVIII o FvW de la coagulación.Pueden ocurrir eventos trombóticos en caso de dosis mayores.
Presentaciones: Caja con un frasco ámpula con liofilizado que contiene 500 UI FVIII/500 UI FvW, y un frasco ámpula con diluyente con 5 ml de agua inyectable con 0.1% de polisorbato 80, una aguja de doble punta, una aguja filtrante y un equipo de infusión.Caja con un frasco ámpula con liofilizado que contiene 1,000 UI FVIII/1,000 UI FvW, y un frasco ámpula con diluyente con 10 ml de agua inyectable con 0.1% de polisorbato 80, una aguja de doble punta, una aguja filtrante y un equipo de infusión.
Recomendaciones sobre almacenamiento: Se almacena a una temperatura entre +2° a +8°C durante 36 meses, o se almacena a una temperatura a no más de +25°C durante 24 meses.No se congele.Protéjase de la luz.
Leyendas de protección: Se ha demostrado la estabilidad de WILATE una vez reconstituido durante 12 horas a temperatura ambiente (máx 25°C), no obstante para evitar la contaminación bacteriana, la solución deberá usarse inmediatamente. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimento. Hecha la mezcla adminístrese de inmediato y deséchese el sobrante. Deséchese las agujas y el equipo de infusión después de su uso. El riesgo de transmisión de agentes infecciosos no puede ser totalmente excluido, cuando se aplican hemoderivados. No se deje al alcance de los niños. Su venta requiere receta médica. Literatura exclusiva para el médico. El plasma humano utilizado en la fabricación de este medicamento es originario de Europa, Estados Unidos de América o México Reporte las sospechas de reacción adversa al correo:farmacovigilancia@co fepris.gob.mx
Nombre y domicilio del laboratorio: Hecho en Austria por: Octapharma Pharmazeutika Produktionsges m.b.H. Oberlaaer StraBe 235 1100 Viena, Austria Acondicionado y distribuido en México por: OCTAPHARMA, S. A. de C. V. Calz. México Tacuba Núm. 1419. Col. Argentina Pte. CP 11230, México, D. F.
Número de registro del medicamento: 195M2009, SSA IV