XELODA®
ROCHE
Denominación genérica: Capecitabina.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: capecitabina 150 mg o 500 mg. Excipiente cbp 1 tableta recubierta.
Indicaciones terapéuticas: Cáncer de mama: XELODA® en combinación con docetaxel está indicado en el tratamiento de pacientes con cáncer de mama metastásico o localmente avanzado, después del fracaso de la quimioterapia citotóxica. La terapia previa pudo haber incluido una antraciclina. XELODA® también está indicado como monoterapia para el tratamiento de pacientes con cáncer de mama metastásico o localmente avanzado, después del fracaso de un esquema de quimioterapia de un taxano y una antraciclina o en quienes no está indicado un esquema de terapia subsecuente con antraciclina. Cáncer colorrectal: XELODA® está indicado como tratamiento adyuvante de pacientes con cáncer de colon. XELODA® está indicado para el tratamiento de pacientes con cáncer colorrectal metastásico. Cáncer gástrico: XELODA® está indicado como tratamiento de primera línea en pacientes con cáncer gástrico avanzado.
Farmacocinética y farmacodinamia: Mecanismo de acción: la capecitabina es un derivado carbamato de la fluoropirimidina, el cual fue diseñado como agente citotóxico tumor-selectivo y tumor-activado, de administración oral. La capecitabina no es citotóxica in vitro. Sin embargo, in vivo, es secuencialmente convertida a la molécula citotóxica 5-fluorouracilo (5-FU), la cual es metabolizada posteriormente. La formación de 5-FU es catalizada preferencialmente en el sitio del tumor, por el factor angiogénico timidina fosforilasa (dThPasa), asociado al tumor, lo que minimiza la exposición de tejidos sanos al 5-FU sistémico. La biotransformación enzimática secuencial de la capecitabina hacia 5-FU, conduce a concentraciones más elevadas dentro de los tejidos tumorales. Tras la administración oral de capecitabina a pacientes con cáncer colorrectal (N=8), la relación entre la concentración de 5-FU en el tumor colorrectal y los tejidos adyacentes fue de 3,2 (con rango de 0,9 a 8,0). La relación entre la concentración de 5-FU en el tumor y el plasma, fue de 21,4 (rango de 3,9 a 59,9), mientras que la relación entre los tejidos sanos y el plasma fue de 8,9 (rango 3,0 hasta 25,8). Se midió la actividad de la timidina fosforilasa y se encontró que es 4 veces mayor en el tumor colorrectal primario que en los tejidos normales adyacentes. Diversos tumores humanos, tales como el de mama, gástrico, colorrectal, cérvicouterino y de ovario, tienen un nivel más elevado de timidina fosforilasa (capaz de convertir el 5'-DFUR [5'-desoxi-5-fluorouridina] a 5-FU), en comparación a los tejidos normales. Las células normales y células tumorales, metabolizan el 5-FU en monofosfato de 5-fluoro-2-desoxiuridina (FdUMP) y trifosfato de 5-fluorouridina (FUTP). Estos metabolitos causan lesión celular por medio de dos mecanismos diferentes. En el primero, el FdUMP y el cofactor N5-10-metilenetetrahidrofolato se unen a la timidilatosintetasa (TS), para formar un complejo ternario de enlace covalente. Esta unión inhibe la formación de timidilato a partir del uracilo. El timidilato es el precursor necesario del trifosfato de timidina, el cual es esencial para la síntesis de ADN, por lo que una deficiencia de este compuesto, puede inhibir la división celular. En el segundo, las enzimas de transcripción nuclear pueden incorporar equivocadamente al FUTP en lugar del trifosfato de uridina (UTP) durante la síntesis del ARN. Este error metabólico puede interferir con los procesos de interpretación del ARN y la síntesis de proteínas. Eficacia: monoterapia - cáncer de colon localmente avanzado: los resultados de un estudio clínico Fase III multicéntrico, aleatorizado en pacientes con cáncer de colon etapa III (Dukes C) respaldan el uso de XELODA® como tratamiento adyuvante para cáncer de colon (estudio XACT: M66001). En este estudio, se asignaron de forma aleatoria a 1.987 pacientes para recibir tratamiento con XELODA® (1.250 mg/m2 dos veces al día por dos semanas seguido de una semana de descanso, administrado en ciclos de 3 semanas durante 24 semanas) o 5-FU y leucovorina (esquema de la Clínica Mayo: 20 mg/m2 de leucovorina IV seguidos de 425 mg/m2 de 5-FU en bolos IV,en los días 1 a 5, cada 28 días, durante 24 semanas). Con XELODA®, la supervivencia sin enfermedad fue al menos equivalente con 5-FU/LV IV (p = 0,0001, 1,2 margen de no inferioridad). En toda la población asignada aleatoriamente, las pruebas para la diferencia de la supervivencia libre de enfermedad y supervivencia global, entre XELODA® y 5-FU/LV, mostraron índices de riesgo de 0.88 (Intervalo de Confianza al 95% 0,77-1,01; p = 0,068) y 0,86 (0,74-1,01; p= 0,060), respectivamente. La media alcanzada en el momento del análisis fue de 6,9 años. Los datos de dos estudios clínicos Fase III, multicéntricos, aleatorizados, comparativos y de idéntico diseño, apoyan el uso de XELODA® como tratamiento de primera línea para el cáncer colorrectal metastásico (SO14695; SO14796). En estos estudios, 603 pacientes fueron seleccionados aleatoriamente para tratamiento con XELODA® (1.250 mg/m2 dos veces al día durante 2 semanas, seguido de un período de descanso de 1 semana, en ciclos de 3 semanas) y 604 pacientes fueron seleccionados aleatoriamente para el tratamiento con 5-FU y leucovorina (esquema de la Clínica Mayo: 20 mg/m2 de leucovorina IV, seguido por 425 mg/m2 de 5-FU IV en bolo, en los días 1 a 5, cada 28 días). Las tasas de respuesta objetiva total en toda la población seleccionada aleatoriamente (evaluación del investigador), fueron del 25.7% con XELODA® frente a 16.7% (esquema de la Clínica Mayo), con una p < 0,0002. El tiempo medio hasta la progresión fue de 140 días para XELODA®, frente a 144 días (esquema de la Clínica Mayo). El promedio de supervivencia fue de 392 días (XELODA®) frente a 391 días (esquema de la Clínica Mayo). Terapia de combinación - tratamiento de primera línea de cáncer de colon y recto metastásico: los datos de un estudio clínico multicéntrico, aleatorio, controlado Fase III (N016966) sustentan el uso de XELODA® en combinación con oxaliplatino o en combinación con oxaliplatino y bevacizumab (BV) para el tratamiento de primera línea de cáncer colorrectal metastásico. El estudio estuvo formado de dos partes: una parte inicial de 2 grupos en donde los pacientes fueron asignados aleatoriamente a dos distintos grupos de tratamiento, incluyendo XELOX o FOLFOX-4, y una subsiguiente parte factorial 2x2 con cuatro diferentes grupos de tratamiento, incluyendo XELOX + placebo (P), FOLFOX-4+P, XELOX+BV, y FOLFOX-4+BV. Los regimenes de tratamiento se resumen en la siguiente tabla.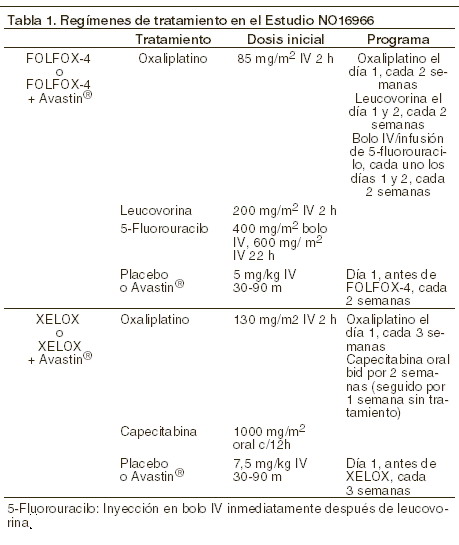
La no inferioridad de los grupos que contenían XELOX comparados con los grupos que contenían FOLFOX-4-en la comparación general se demostró en términos de la supervivencia libre de progresión en la población de pacientes elegibles y en la población con intención de tratamiento (ITT por sus siglas en inglés) (refiérase a la tabla a continuación). Los resultados indican que XELOX es equivalente a FOLFOX-4 en términos de supervivencia global. Una comparación de XELOX más bevacizumab contra FOLFOX-4 más bevacizumab fue un análisis exploratorio especificado previamente. En esta comparación del subgrupo de tratamiento, XELOX más bevacizumab fue similar comparado con FOLFOX-4 más bevacizumab en términos de supervivencia libre de progresión (índice de riesgo 1,01 [Intervalo de Confianza al 97,5%: 0,84, 1,22]). El seguimiento promedio al momento de los análisis primarios en la población con intención de tratamiento fue de 1,5 años; los datos de los análisis después de 1 año adicional de seguimiento también se incluyen en la siguiente tabla.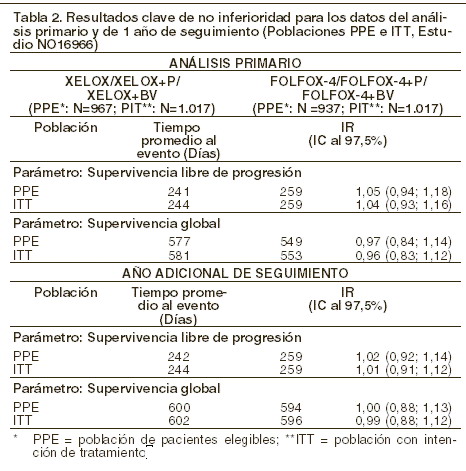
Los datos de un estudio aleatorio, controlado, Fase III (CAIRO) sustentan el uso de XELODA® a una dosis inicial de 1.000 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan para el tratamiento de primera línea de pacientes con cáncer colorrectal metastásico. La eficacia en términos del Indice de Respuesta Global (IRG), Supervivencia Libre de Progresión (SLP) y Supervivencia Global (SG) fue similar a la reportada en estudios pivote de 5-FU, leucovorina, e irinotecan (FOLFIRI). Los datos de un análisis interino de un estudio multicéntrico, aleatorio, controlado, Fase II (AIO KRK 0604) sustentan el uso de XELODA® a una dosis inicial de 800 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan y bevacizumab para el tratamiento de pacientes con cáncer colorrectal metastásico. 115 pacientes fueron asignados aleatoriamente al tratamiento con XELODA® combinado con irinotecan (XELIRI) y bevacizumab: XELODA® (800 mg/m2 dos veces al día por dos semanas seguido por un periodo de descanso de 7 días), irinotecan (200 mg/m2 como infusión de 30 minutos el día 1 cada 3 semanas) y bevacizumab (7,5 mg/kg como infusión de 30 a 90 minutos el día 1 cada 3 semanas); un total de 118 pacientes fueron asignados aleatoriamente al tratamiento con XELODA® en combinación con oxaliplatino más bevacizumab: XELODA® (1.000 mg/m2 dos veces al día por dos semanas seguido por un periodo de descanso de 7 días), oxaliplatino (130 mg/m2 como infusión de 2 horas el día 1 cada 3 semanas) y bevacizumab (7,5 mg/m2 como infusión de 30 a 90 minutos el día 1 cada 3 semanas). La supervivencia libre de progresión a 6 meses en la población con intención de tratamiento fue del 80% (XELIRI más bevacizumab) contra 74% (XELOX más bevacizumab). El índice de respuesta global (respuesta completa más respuesta parcial) fue de 47% (XELIRI más bevacizumab) contra 45% (XELOX más bevacizumab). Terapia de combinación - tratamiento de segunda línea de cáncer colorrectal: los datos de un estudio clínico multicéntrico, aleatorio, controlado, Fase III (NO16967) sustentan el uso de XELODA® en combinación con oxaliplatino para el tratamiento de segunda línea de cáncer colorrectal metastásico. En este estudio, 627 pacientes con cáncer colorrectal metastásico que habían recibido tratamiento previo con irinotecan en combinación con un régimen de fluoropirimidina como terapia de primera línea fueron asignados aleatoriamente al tratamiento con XELOX o FOLFOX-4. Para el esquema de dosificación de XELOX y FOLFOX-4 (sin adición de placebo o bevacizumab), refiérase a la tabla 1. Se demostró que XELOX no es inferior a FOLFOX-4 en términos de supervivencia libre de progresión en la población conforme al protocolo y en la población con intención de tratamiento (ver la tabla a continuación). Los resultados indican que XELOX es equivalente a FOLFOX-4 en términos de supervivencia global. El seguimiento promedio al momento de los análisis primarios en la población con intención de tratamiento fue de 2,1 años; los datos de los análisis después de un seguimiento adicional de 6 meses también se incluyen en la siguiente tabla.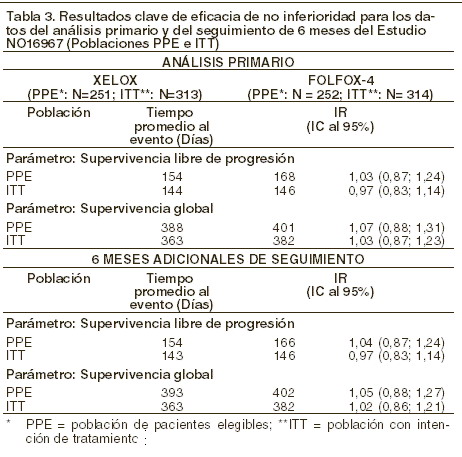
Un análisis conjunto de los datos de eficacia del tratamiento de primera línea (estudio NO16966; parte inicial de 2 grupos) y del tratamiento de segunda línea (estudio NO16967) sustenta aún más los resultados de no inferioridad de XELOX contra FOLFOX-4 según lo obtenido en los estudios individuales: supervivencia libre de progresión en la población conforme a protocolo (índice de riesgo 1.00 [IC al 95%: 0,88; 1,14]) con una supervivencia libre de progresión promedio de 193 días (XELOX; 508 pacientes) contra 204 días (FOLFOX-4; 500 pacientes). Los resultados indican que XELOX es equivalente a FOLFOX-4 en términos de supervivencia global (índice de riesgo 1.01 [IC al 95%: 0.87; 1.17]) con una supervivencia global promedio de 468 días (XELOX) contra 478 días (FOLFOX-4). Terapia en combinación - cáncer gástrico: los datos de un estudio clínico Fase III, multicéntrico, aleatorizado, controlado en pacientes con cáncer gástrico metastásico o avanzado, apoyan el uso de XELODA® para el tratamiento de primera línea de cáncer gástrico avanzado. En este estudio, 160 pacientes fueron asignados aleatoriamente al tratamiento con XELODA® (1000 mg/m2 dos veces al día, durante 2 semanas seguido por un periodo de descanso de 7 días) y cisplatino (80 mg/m2 como una infusión durante 2 horas, cada 3 semanas). Un total de 156 pacientes fueron asignados aleatoriamente al tratamiento con 5-FU (800 mg/m2 por día, infusión continua en los días 1 a 5, cada 3 semanas) y cisplatino (80 mg/m2 como infusión durante 2 horas en el día 1, cada 3 semanas). El objetivo primario del estudio se cumplió, XELODA® en combinación con cisplatino fue al menos equivalente al 5-FU en combinación con cisplatino, en términos de supervivencia libre de progresión según el protocolo. El resultado de la duración de la supervivencia (supervivencia total) fue similar al resultado de la supervivencia libre de progresión (ver tabla abajo).
Los datos de un estudio multicéntrico, aleatorio, Fase III comparando la capecitabina al 5-FU y oxaliplatino a cisplatino en pacientes con cáncer gástrico avanzado sustentan el uso de XELODA® para el tratamiento de primera línea de cáncer gástrico avanzado. En este estudio, 1002 pacientes fueron asignados aleatoriamente en un diseño factorial 2x2 a uno de los siguientes 4 grupos: ECF: epirubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), cisplatino (60 mg/m2 como infusión de dos horas el día 1 cada 3 semanas) y 5-FU (200 mg/m2 diariamente administrado mediante infusión continua vía línea central). ECX: epirubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), cisplatino (60 mg/m2 como infusión por dos horas el día 1 cada 3 semanas) y XELODA® (625 mg/m2 dos veces al día de manera continua). EOF: epirubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como infusión de 2 horas el día 1 cada tres semanas) y 5-FU (200 mg/m2 diaria administrado por infusión continua vía línea central). EOX: epirubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como infusión de 2 horas el día 1 cada tres semanas) y XELODA® (625 mg/m2 dos veces al día de manera continua). Los análisis de eficacia primaria en la población conforme al protocolo, demostraron la no inferioridad en la supervivencia global para la capecitabina - contra los regímenes a base de 5-FU - (índice de riesgo 0,86, IC al 95%: 0,8 a 0,99) y para oxaliplatino - contra regímenes a base de cisplatino (índice de riesgo 0,92, IC al 95%: 0,8 a 1,1). La supervivencia global promedio fue de 10,9 meses en los regímenes a base de capecitabina y de 9,6 meses en los regímenes a base de 5-FU. La supervivencia global promedio fue de 10,0 meses en los regímenes a base de cisplatino y de 10.4 meses en los regímenes a base de oxaliplatino. XELODA® también se ha usado en combinación con oxaliplatino para el tratamiento de cáncer gástrico avanzado. Estudios con monoterapia de XELODA® indican que XELODA® tiene actividad en el cáncer gástrico avanzado. Cáncer de colon, colorrectal y gástrico avanzado: meta-análisis: un meta-análisis de seis estudios clínicos (estudios SO14695, SO14796, M66001, NO16966, NO16967, M17032) sustentan el reemplazo de 5-FU por XELODA® en el tratamiento de un solo fármaco y de combinación en el cáncer gastrointestinal. El análisis conjunto incluyó a 3097 pacientes tratados con regímenes que contenían XELODA® y a 3074 pacientes tratados con regímenes que contenían 5-FU. El índice de riesgo para la supervivencia global fue de 0.96 (IC al 95%: 0.90; 1.02) indicando que los regímenes que contienen XELODA® son equivalentes a los regímenes que contienen 5-FU. Terapia de combinación - cáncer de mama: los datos de un estudio clínico multicéntrico, aleatorio, controlado, Fase III sustentan el uso de XELODA® en combinación con docetaxel para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastásico después de la falla de la quimioterapia citotóxica, incluyendo una antraciclina. En este estudio, 255 pacientes fueron asignados aleatoriamente al tratamiento con XELODA® (1.250 mg/m2 dos veces al día por 2 semanas seguido por un periodo de descanso de 1 semana) y docetaxel (75 mg/m2 como infusión intravenosa de 1 hora cada 3 semanas). Un total de 256 pacientes fueron asignados aleatoriamente al tratamiento con docetaxel solo (100 mg/m2 como infusión intravenosa por 1 hora cada 3 semanas). La supervivencia fue superior en el grupo de combinación de XELODA® + docetaxel (p = 0,0126). La supervivencia promedio fue de 442 días (XELODA® + docetaxel) contra 352 días (docetaxel solo). Los índices de respuesta objetiva general en toda la población asignada aleatoriamente (evaluación del investigador) fueron de 41.6% (XELODA® + docetaxel) contra 29.7% (docetaxel solo); p = 0,0058. El tiempo para la progresión de la enfermedad o muerte fue superior en el grupo de combinación XELODA® + docetaxel (p < 0,0001). El tiempo promedio a la progresión fue de 186 días (XELODA® + docetaxel) contra 128 días (docetaxel solo). Monoterapia - cáncer de mama: los datos de dos estudios clínicos multicéntricos Fase II sustentan el uso de la monoterapia con XELODA® para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastásico después de la falla de un régimen de taxano y una quimioterapia que contenga antraciclinas o para quienes no esté indicada la terapia con antraciclinas. En este estudio, un total de 236 pacientes fueron tratados con XELODA® (1250 mg/m2 dos veces al día por 2 semanas seguido por un periodo de descanso de 1 semana). Los índices de respuesta objetiva general (evaluación del investigador) fueron del 20% (primer estudio) y 25% (segundo estudio). El tiempo promedio para la progresión fue de 93 y 98 días. La supervivencia promedio fue de 384 y 373 días. Farmacocinética: absorción: Después de la administración oral, la capecitabina se absorbe rápida y en gran cantidad, seguido de una amplia conversión hacia los metabolitos 5'-deoxi-5-fluorocitidina (5'-DFCR) y 5'-DFUR. La administración con los alimentos disminuye la velocidad de absorción de la capecitabina, pero tiene solo efectos mínimos en las áreas bajo la curva (ABC) del 5'-DFUR y el metabolito subsecuente 5-FU. En la dosis de 1250 mg/m2 en el día 14 con administración, posterior a la ingesta de alimentos, las concentraciones máximas en plasma (Cmáx, en mg/ml) de la capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL, fueron de 4.47, 3.05, 12.1, 0.95 y 5.46, respectivamente. Los tiempos en alcanzarse las concentraciones máximas en plasma (Tmáx, en horas), fueron de 1,50, 2,00, 2,00, 2,00 y 3,34 respectivamente. Los valores del ABC0-inf, en mgh/ml, fueron 7,75, 7,24, 24,6, 2,03 y 36,3 respectivamente. Distribución: unión a proteínas: los estudios realizados in vitro en plasma humano, han determinado que la capecitabina, 5'-DFCR, 5'-DFUR y 5-FU se unen a las proteínas en un 54%, 10%, 62% y 10% respectivamente, principalmente a la albúmina. Metabolismo: la capecitabina es metabolizada primero por la carboxilesterasa hepática en 5'-DFCR, el cual a su vez es convertido a 5'-DFUR por la citidina desaminasa principalmente localizada en el hígado y los tejidos tumorales. La formación de 5-FU se produce de forma preferencial en el sitio del tumor, por el factor angiogénico asociado al tumor, dThdPasa, minimizando así la exposición de los tejidos corporales sanos a la exposición sistémica del 5-FU. El ABC plasmático del 5-FU es de 6 a 22 veces menor que el producido después de la administración intravenosa en bolo de 5-FU (dosis de 600 mg/m2). Los metabolitos de capecitabina se convierten en citotóxicos únicamente después de la conversión hacia 5-FU y los anabolitos de 5-FU (ver Mecanismo de acción). El 5-FU es posteriormente catabolizado hacia los metabolitos inactivos dihidro-5-fluorouracilo (FUH2), ácido 5-fluoro-ureidopropiónico (FUPA) y a-fluoro-b-alanina (FBAL), a través de la dihidropirimidina deshidrogenasa (DPD), lo cual es limitante de la velocidad. Eliminación: la vida media de eliminación (t½, en horas) de la capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL, fue de 0,85, 1,11, 0,66, 0,76 y 3,23 horas, respectivamente. La farmacocinética de la capecitabina ha sido evaluada dentro del rango de dosis de 502 a 3.514 mg/m2/día. Los parámetros farmacocinéticos de la capecitabina, 5'-DFCR y 5'-DFUR medidos en los días 1 y 14, fueron similares. El ABC del 5-FU fue de 30% - 35% más elevado en el día 14, pero no se incrementó subsecuentemente (día 22). A dosis terapéuticas, las farmacocinéticas de la capecitabina y sus metabolitos, fueron proporcionales a la dosis, excepto para el 5-FU. Después de la administración oral, los metabolitos de la capecitabina son principalmente recuperados en la orina. La mayor parte (95,5%) de la dosis administrada de capecitabina, se recupera en la orina. La excreción fecal es mínima (2,6%). El principal metabolito excretado en la orina es el FBAL, el cual representa el 57% de la dosis administrada. Aproximadamente el 3% de la dosis administrada se excreta en la orina en forma inalterada. Terapia de combinación: los estudios Fase I que evaluaron el efecto de XELODA® sobre la farmacocinética ya sea de docetaxel o paclitaxel y viceversa, no mostraron efectos inducidos por la administración de XELODA®, sobre la farmacocinética de estos fármacos (Cmáx y ABC), ni efectos del docetaxel o paclitaxel sobre la farmacocinética del 5'-DFUR (el metabolito más importante de la capecitabina). Farmacocinética en poblaciones especiales: se realizó un análisis farmacocinético poblacional, después del tratamiento con XELODA® a 505 pacientes con cáncer colorrectal, quienes recibieron la dosis de 1.250 mg/m2 dos veces al día. El sexo, presencia o ausencia de metástasis hepáticas basales, el estado físico según la escala de Karnofsky, la bilirrubina total, la albúmina sérica, las concentraciones séricas de ASAT y ALAT, no tuvieron efecto estadísticamente significativo sobre la farmacocinética del 5'-DFUR, 5-FU y FBAL. Pacientes con insuficiencia hepática debida a metástasis en el hígado: no se observaron efectos clínicamente significativos sobre la bioactivación y farmacocinética de capecitabina, en pacientes con cáncer que presentaban insuficiencia hepática leve a moderada, debida a metástasis en el hígado (ver Instrucciones especiales de dosificación). No existen datos farmacocinéticos en pacientes con insuficiencia hepática severa. Pacientes con insuficiencia renal: con base en un estudio de farmacocinética realizado en pacientes con cáncer que presentaban insuficiencia renal leve a severa, se determinó que no existe evidencia de efectos sobre la depuración de creatinina en la farmacocinética de la capecitabina intacta y del 5-FU. Sin embargo, se encontró que la depuración de creatinina influye en la exposición sistémica del 5-DFUR (35% de incremento en el ABC cuando la depuración de creatinina disminuye en 50%) y del FBAL (114% de incremento en el ABC cuando la depuración de creatinina disminuye en 50%). El FBAL es un metabolito sin actividad antiproliferativa, mientras que el 5'-DFUR es el precursor directo del 5-FU (ver Instrucciones especiales de dosificación). Ancianos: con base en un estudio de farmacocinética poblacional que incluyó a pacientes con un amplio rango de edad (27 a 86 años) de los cuales 234 (46%) eran pacientes con 65 años o más, se observó que la edad no tuvo influencia en la farmacocinética del 5'-DFUR y 5-FU. Sin embargo el ABC del FBAL se incrementó con la edad (20% de incremento en la edad resulta en un 15% de incremento en el ABC del FBAL). Este incremento es probablemente debido al cambio en la función renal que se produce con la edad (ver Instrucciones especiales de dosificación y Farmacocinética en poblaciones especiales). Raza: en un análisis de farmacocinética poblacional de 455 pacientes de raza blanca (90,1%), 22 pacientes de raza negra (4,4%) y 28 pacientes de otra raza o etnia (5,5%), se determinó que la farmacocinética de los pacientes negros no fue diferente a la de los pacientes blancos ni de otras razas.
Contraindicaciones: XELODA® está contraindicado en pacientes con hipersensibilidad conocida a la capecitabina o a cualquiera de sus componentes. XELODA® está contraindicado en pacientes que tienen antecedentes de reacciones adversas graves o inesperadas a la terapia con fluoropirimidinas o con hipersensibilidad conocida al fluorouracilo. Al igual que con otras fluoropirimidinas, XELODA® está contraindicado en pacientes con deficiencia conocida de deshidrogenasa de dihidropirimidina (DPD). XELODA® no debe ser administrado concomitantemente con sorivudina o sus análogos químicamente relacionados, tales como brivudina (ver Interacciones medicamentosas y de otro género). XELODA® está contraindicado en pacientes con insuficiencia renal grave (depuración de creatinina por debajo de 30 ml/min). Si existen contraindicaciones a algunos de los agentes en un régimen de combinación, el agente no deberá ser utilizado.
Precauciones generales: Generalidades: los pacientes tratados con XELODA® deben ser cuidadosamente monitoreados para vigilar la aparición de manifestaciones de toxicidad. La mayoría de los eventos adversos son reversibles y no se requiere de la suspensión permanente de la terapia, aún cuando puede ser necesario aplazar o reducir las dosis (ver Dosis y vía de administración). El espectro de cardiotoxicidad observado con XELODA® es similar al de otras pirimidinas fluorinadas. Esto incluye infarto del miocardio, angina de pecho, arritmias, paro cardíaco, insuficiencia cardíaca y cambios electrocardiográficos. Estos eventos adversos pueden ser más comunes en pacientes con antecedentes de enfermedad arterial coronaria. Rara vez, los efectos de toxicidad severa (tales como estomatitis, diarrea, neutropenia y neurotoxicidad) asociados con el 5-fluorouracilo, han sido atribuidos a la deficiencia de dihidropirimidina deshidrogenasa (DPD). Por lo tanto, no puede descartarse completamente una relación entre la disminución de los niveles de DPD y el aumento de los efectos tóxicos potencialmente fatales del 5-fluorouracilo. XELODA® puede inducir síndrome mano-pie (eritrodisestesia palmo-plantar o eritema acral inducido), el cual es una toxicidad cutánea. Para los pacientes con enfermedad metastásica que recibieron monoterapia con XELODA®, el tiempo promedio para el inicio del evento fue de 79 días (rango de 11 a 360 días), con un rango de severidad que estuvo entre los grados 1 a 3. El grado 1 del síndrome mano-pie se define como entumecimiento, disestesia/parestesia, hormigueo o eritema de manos y/o pies y/o malestar que no interfiere con las actividades normales. El grado 2 se define como la aparición de inflamación y eritema doloroso de manos y/o pies y/o malestar que afecta las actividades diarias del paciente. El grado 3 se define como la descamación húmeda, ulceración, ampollas o dolor intenso de manos y/o pies, y/o malestar severo que hace que el paciente no sea capaz de trabajar o realizar sus actividades diarias. Si se presenta síndrome mano-pie grado 2 o 3, deberá interrumpirse la administración de XELODA®, hasta que el evento se resuelva o disminuya en intensidad hasta el grado 1. Después de la aparición de síndrome mano-pie grado 3, las dosis subsecuentes de XELODA® deberán disminuirse (ver Dosis y vía de administración). Cuando se utiliza XELODA® y cisplatino en combinación, no se recomienda el uso de la vitamina B6 (piridoxina) como tratamiento sintomático o profiláctico secundario del síndrome mano-pie, ya que, de acuerdo con reportes publicados, puede disminuir la eficacia del cisplatino. XELODA® puede inducir hiperbilirrubinemia. La administración de XELODA® deberá interrumpirse si se presentan incrementos en los niveles de bilirrubina, relacionados con el tratamiento, mayores de 3,0 veces por arriba del límite superior de normalidad (LSN) o se producen elevaciones en las aminotransferasas hepáticas relacionadas con el tratamiento (ALAT, ASAT), 2,5 veces arriba del LSN. El tratamiento puede ser reanudado cuando los niveles de bilirrubina disminuyan a ≤ 3,0 x LSN o los niveles de aminotransferasas hepáticas disminuyan a ≤ 2,5 x LSN. En un estudio de interacción de medicamentos, con la administración de una dosis única de warfarina, se observó un incremento significativo en el promedio del ABC (+57%) de la warfarina-S. Estos resultados sugieren una interacción, probablemente debida a la inhibición del sistema de la isoenzima 2C9 del citocromo P450, inducida por la capecitabina. Los pacientes que reciben terapia con XELODA® y una terapia anticoagulante concomitante con derivados de la cumarina (INR o tiempo de protrombina) deben ser vigilados cuidadosamente con pruebas de coagulación y ajuste de la dosis del anticoagulante de acuerdo a los resultados (ver Interacciones medicamentosas y de otro género). En pacientes con cáncer colorrectal metastásico entre 60 a 79 años de edad, que recibieron monoterapia con XELODA®, la incidencia de toxicidad gastrointestinal fue similar a la de la población general. Un porcentaje mayor de pacientes con 80 años de edad o más, experimentó eventos adversos gastrointestinales grado 3 o 4 reversibles, tales como diarrea, náusea y vómito (ver Instrucciones especiales de dosificación). Un análisis de los datos de seguridad en pacientes con 60 años de edad o más, tratados con la terapia combinada de XELODA® más docetaxel, mostró un aumento en la incidencia de eventos adversos grado 3 y 4 relacionados con el tratamiento, así como de eventos adversos graves relacionados con el tratamiento y retiros tempranos del tratamiento debidos a eventos adversos, en comparación con los pacientes menores de 60 años de edad. Los pacientes con insuficiencia hepática deben ser cuidadosamente monitoreados cuando se administre XELODA®. Se desconoce el efecto de la insuficiencia hepática no debida a metástasis en hígado o de la insuficiencia hepática grave, sobre el metabolismo de XELODA® (ver Farmacocinética e Instrucciones especiales de dosificación). Los médicos deben tener cuidado cuando se administre XELODA® a pacientes con alteración de la función renal. Como se ha observado con el 5-fluorouracilo, la incidencia de eventos adversos grado 3 o 4 relacionados con el tratamiento, es más elevada en pacientes con insuficiencia renal moderada (depuración de creatinina 30-50 ml/min). Advertencias: diarrea: XELODA® puede inducir diarrea, la cual en algunas ocasiones puede ser severa. Los pacientes con diarrea severa deben ser cuidadosamente monitoreados y, si se deshidratan, deberán recibir líquidos y reemplazo de electrolitos. Deberá iniciarse el tratamiento con antidiarréicos convencionales (por ejemplo loperamida), según esté médicamente indicado, tan pronto como sea posible. La reducción de dosis deberá ser aplicada como sea necesaria. Deshidratación: se debe prevenir la deshidratación o corregirla a su aparición. Los pacientes con anorexia, astenia, náusea, vómito o diarrea pueden deshidratarse más rápidamente. Si se presenta deshidratación grado 2 (o mayor), se deberá interrumpir el tratamiento con XELODA® inmediatamente y corregir la deshidratación. El tratamiento no se deberá reiniciar hasta que el paciente haya sido rehidratado y se haya corregido o controlado cualquier causa que la haya precipitado. Se deben aplicar las modificaciones a las dosis para la reacción adversa precipitante conforme sea necesario (ver Dosis y vía de administración).
Restricciones de uso durante el embarazo y la lactancia: Embarazo: embarazo categoría D. No existen estudios clínicos de XELODA® realizados en mujeres embarazadas; sin embargo, con base en las propiedades farmacológicas y toxicológicas de XELODA®, puede asumirse que XELODA® puede causar daño fetal si se administra a mujeres embarazadas. En los estudios de toxicidad reproductiva en animales, la administración de capecitabina fue teratógena y letal para el embrión. Estos hallazgos son efectos esperados de los derivados de las fluoropirimidinas. La capecitabina debe ser considerada como un teratógeno potencial para el humano. XELODA® no debe utilizarse durante el embarazo. Si se administra XELODA® durante el embarazo o si la paciente se embaraza mientras recibe este fármaco, se le deberá advertir sobre el riesgo potencial para el feto. A las mujeres con potencial de concebir, se les debe aconsejar evitar el embarazo mientras se encuentran en tratamiento con XELODA®. Lactancia: se desconoce si el fármaco es excretado en la leche humana. En un estudio de administración oral única de XELODA® a ratonas en período de lactancia, se detectó una cantidad importante de los metabolitos de capecitabina en la leche. Se deberá suspender la lactancia durante el tratamiento con XELODA®.
Reacciones secundarias y adversas: Experiencia de los estudios clínicos: las reacciones adversas a medicamento (RAM) que el investigador consideró estar posiblemente, probablemente o remotamente relacionas con la administración de XELODA® se han obtenido de los estudios clínicos realizados con la monoterapia con XELODA® (en la terapia adyuvante del cáncer de colon, en cáncer colorrectal metastásico y cáncer de mama metastásico), y de estudios clínicos realizados con XELODA® en combinación con docetaxel (cáncer de mama metastásico), con cisplatino (cáncer gástrico avanzado), o en combinación de oxaliplatino con o sin bevacizumab (cáncer colorrectal metastásico). Monoterapia con XELODA®: los datos de seguridad de la monoterapia de XELODA® se reportaron para pacientes que recibieron tratamiento adyuvante para el cáncer de colon y para los pacientes que recibieron tratamiento para cáncer de mama metastásico o cáncer colorrectal metastásico. La información de seguridad incluye datos de un estudio Fase III en cáncer de colon adyuvante (995 pacientes tratados con XELODA® y 974 tratados con 5-FU/LV IV) y de cuatro estudios Fase II en pacientes de sexo femenino con cáncer de mama (N = 319) y 3 estudios (1 estudio Fase II y 2 Fase III) en pacientes de sexo femenino y masculino con cáncer colorrectal (N = 630). El perfil de seguridad de la monoterapia de XELODA® es comparable en pacientes que recibieron tratamiento adyuvante para cáncer de colon y en aquellos que recibieron tratamiento para cáncer de mama metastásico o cáncer colorrectal metastásico. La intensidad de RAM se evaluó conforme a las categorías de toxicidad del sistema de evaluación del NCIC CTC.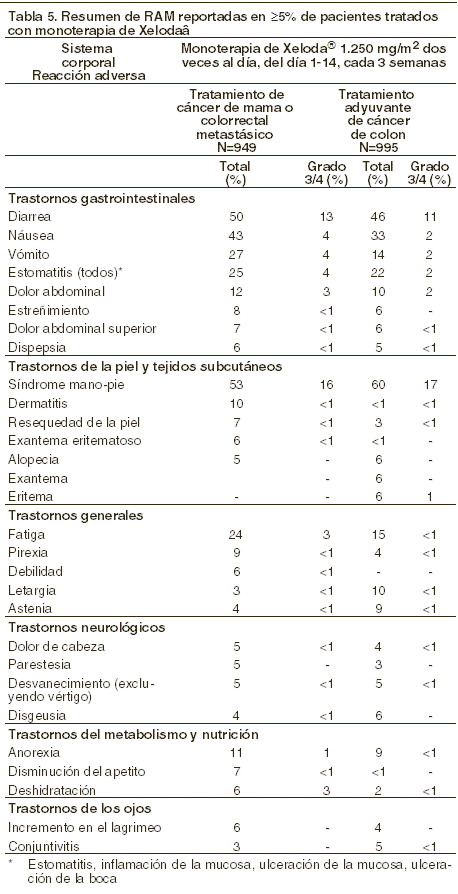
Las fisuras cutáneas fueron reportadas como por lo menos remotamente relacionadas con XELODA®, en menos del 2% de los pacientes, en siete estudios clínicos concluidos (N=949). Las siguientes RAM´s (Reacciones adversas a medicamento) representan toxicidades conocidas de la terapia con la administración de fluoropirimidinas y se reportaron como por lo menos remotamente relacionados con XELODA®, en menos del 5% de los pacientes de 7 estudios concluidos (N=949): Gastrointestinales: boca seca, flatulencia, heces líquidas, RAM´s relacionadas a inflamación/ulceración de las membranas mucosas tales como esofagitis, gastritis, duodenitis, colitis y hemorragia gastrointestinal. Cardíacos: edema de las extremidades inferiores, dolor torácico de origen cardíaco, incluyendo angina, cardiomiopatía, isquemia/infarto del miocardio, insuficiencia cardíaca, muerte súbita, taquicardia, arritmias auriculares incluyendo fibrilación auricular y extrasístoles ventriculares. Neurológicos: disgeusia, insomnio, confusión, encefalopatía y signos cerebelares tales como ataxia, disartria, alteración del equilibrio y coordinación anormal. Infecciones: eventos relacionados con depresión de la médula ósea, inmunosupresión y/o afección de membranas mucosas tales como infecciones locales y sistémicas fatales (incluyendo etiología bacteriana, viral y micótica) y sepsis. Hematológicos: anemia, depresión de la médula ósea (notificada como RAM) y pancitopenia. Dermatológicos: prurito, exfoliación localizada, hiperpigmentación cutánea, trastornos de las uñas, reacciones de fotosensibilidad, síndrome postradiación, onicolisis, uñas frágiles, decoloración y/o distrofia de las uñas. Síntomas generales: astenia, dolor en extremidades, letargo y dolor pectoral. Oculares: conjuntivitis e irritación ocular. Respiratorios: disnea y tos. Musculoesqueléticos: dorsalgia, mialgia y artralgia. Trastornos psiquiátricos: depresión. Durante los estudios clínicos y en la exposición postcomercialización, se han reportado insuficiencia hepática y hepatitis colestásica. No se ha establecido la relación causal con el tratamiento de XELODA®. Terapia en combinación con XELODA®- cáncer de mama: combinación de XELODA® y docetaxel: los efectos indeseables asociados a la monoterapia con XELODA® pueden presentarse también cuando se utiliza XELODA® en combinación con docetaxel. En la siguiente tabla, se describen los efectos indeseables relacionados al tratamiento, reportados con mayor frecuencia (≥ 5%) en un estudio Fase III, realizado en pacientes con cáncer de mama que tuvieron fracaso al tratamiento con antraciclinas. También se presentan los efectos indeseables relacionados con el tratamiento descrito en el grupo de referencia de este estudio, utilizando la dosis habitual de docetaxel. La intensidad de las reacciones adversas fue graduada de acuerdo a la toxicidad en categorías de NCIC CTC sistema de graduación.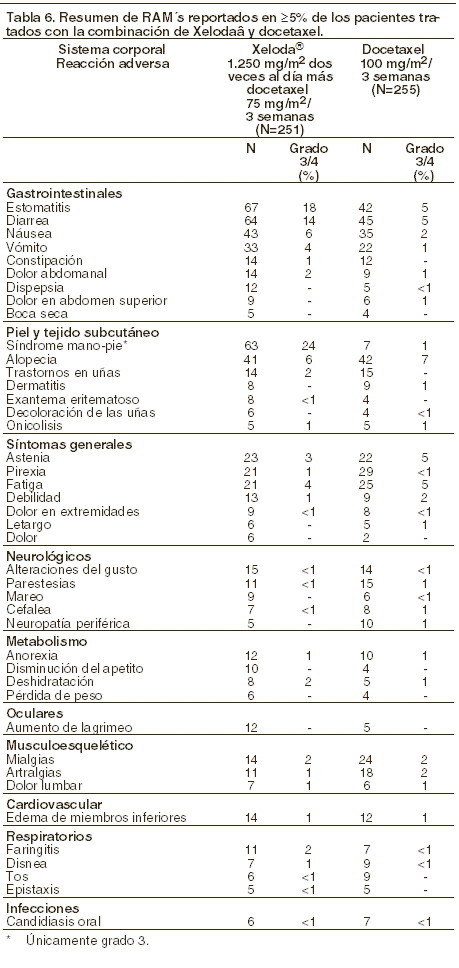
XELODA® terapia combinada - cáncer gástrico: combinación de XELODA® y cisplatino: en la siguiente tabla, se presentan las RAM´s más frecuentes (≥ 5%) relacionados con el tratamiento de pacientes con cáncer gástrico avanzado, reportados en un estudio Fase III. También se presentan las RAM´s relacionados al tratamiento, reportados en el brazo de comparación de este estudio, utilizando 5-FU y cisplatino.07.JPG">
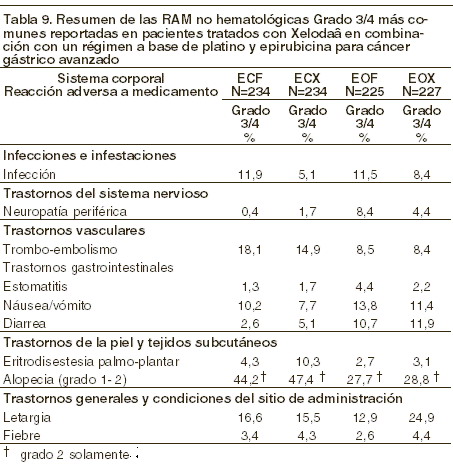
Las RAM´s raras o no comunes reportadas con la terapia de combinación de XELODA® más cisplatino, son consistentes con las RAM´s reportadas para la monoterapia con XELODA® o la monoterapia con cisplatino (ver la información para prescribir de cisplatino). XELODA® y oxaliplatino en combinación: el perfil de seguridad de XELODA® en combinación con oxaliplatino para el tratamiento de cáncer gástrico avanzado es comparable al de pacientes que recibieron XELODA® en combinación con oxaliplatino para el tratamiento de cáncer colorrectal metastásico. XELODA® en combinación con un régimen a base de platino y epirubicina para cáncer gástrico avanzado: las RAM grado 3/4 más comunes reportadas en pacientes tratados con XELODA® en combinación con un régimen a base de platino y epirubicina para cáncer gástrico avanzado se presentan en las siguientes tablas. También se presentan las RAM reportadas en el grupo de comparación de este estudio, usando 5-FU en combinación con un régimen a base de platino y epirubicina.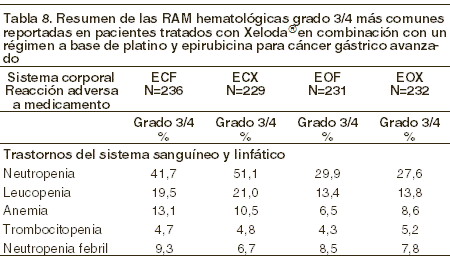
Terapia de combinación de XELODA® - cáncer colorrectal: XELODA® en combinación con oxaliplatino: la siguiente tabla muestra las RAM más frecuentes (≥ 5%) reportadas en pacientes con cáncer colorrectal metastásico que recibieron el tratamiento de primera línea (Estudio NO16966) o el de segunda línea (Estudio NO16967) con XELODA® en combinación con oxaliplatino (XELOX). En el Estudio NO16966, la comparación conjunta de XELOX contra FOLFOX-4 incluye los datos de seguridad conjuntos del grupo de XELOX de la parte inicial de 2 grupos del estudio y del grupo de XELOX + placebo (P) de la parte factorial 2x2 del estudio contra los datos de seguridad conjuntos del grupo de FOLFOX-4 de la parte inicial de 2 grupos del estudio y del grupo FOLFOX-4+P de la parte factorial 2x2 del estudio (ver Eficacia). La intensidad de reacciones adversas se evalúa conforme a las categorías de toxicidad del sistema de evaluación del NCI CTCAE versión 3,0.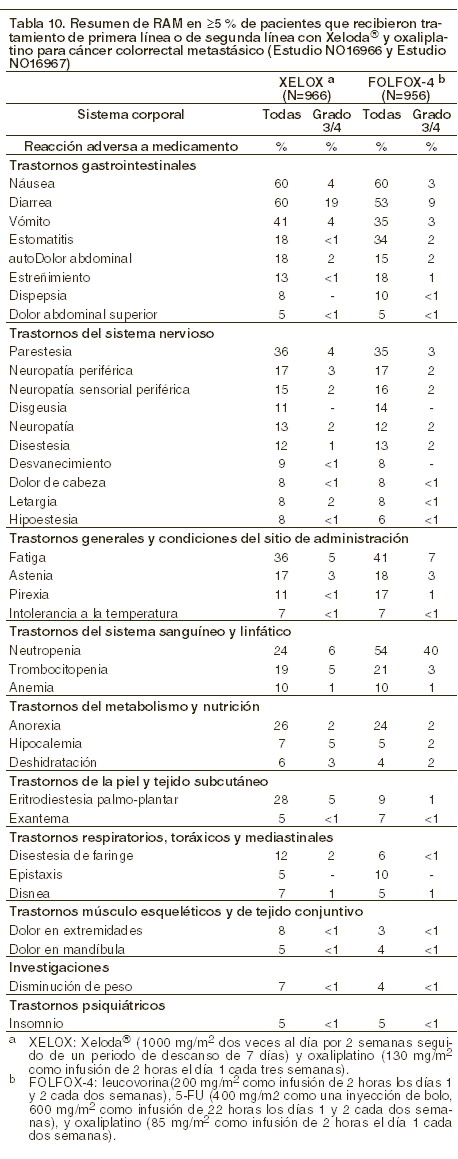
Las RAM raras o poco comunes reportadas para la combinación de XELODA® con oxaliplatino son consistentes con las RAM reportadas para la monoterapia de XELODA® o la monoterapia de oxaliplatino (refiérase a la información de prescripción de oxaliplatino). XELODA® en combinación con oxaliplatino y bevacizumab: la siguiente tabla muestra las RAM más frecuentes (≥ 5%) reportadas en un estudio Fase III (NO16966) de pacientes con cáncer colorrectal metastásico que recibieron tratamiento de primera línea con XELODA® en combinación con oxaliplatino y bevacizumab (XELOX+BV). La comparación de XELOX+BV contra FOLFOX-4+BV incluye datos de seguridad del grupo de XELOX+BV y al grupo de FOLFOX-4+BV de la parte factorial 2x2 del estudio. La intensidad de las reacciones adversas se evaluó conforme a las categorías de toxicidad del sistema de evaluación del NCI CTCAE versión 3.0.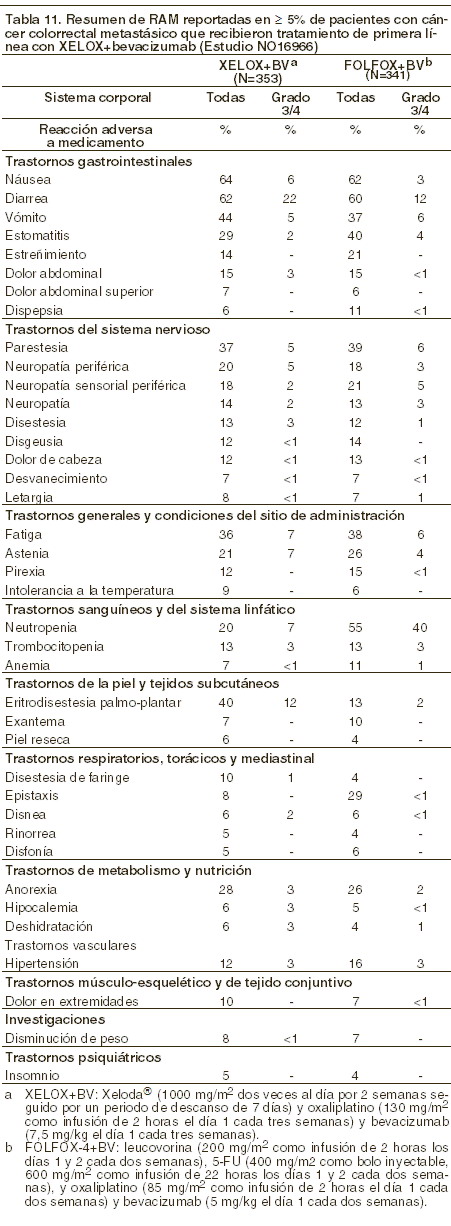
Las RAM raras o poco comunes reportadas para la combinación de XELODA® con oxaliplatino y bevacizumab son consistentes con las RAM reportadas para la monoterapia de XELODA® o la monoterapia de oxaliplatino o la terapia de combinación de bevacizumab (refiérase a la información de prescripción de oxaliplatino o bevacizumab).
XELODA® en combinación con irinotecan con o sin bevacizumab: la siguiente tabla muestra las RAM grado 3/4 más comunes reportadas en pacientes que recibieron el tratamiento de primera línea con XELODA® en combinación con irinotecan con o sin bevacizumab para cáncer colorrectal metastásico.
Experiencia postcomercialización: se han identificado las siguientes reacciones adversas durante el uso postcomercialización: muy raras: estenosis del conducto lagrimal. Muy raras: se ha reportado insuficiencia hepática y hepatitis colestásica durante los estudios clínicos y en la exposición posterior a la comercialización.
Interacciones medicamentosas y de otro género: Anticoagulantes cumarínicos: se ha reportado alteración de los parámetros de coagulación y/o sangrado, en pacientes que tomaron XELODA® concomitantemente con anticoagulantes derivados de la cumarina, tales como la warfarina y el fenprocumón. Estos eventos adversos se presentaron en pocos días y hasta varios meses después del inicio del tratamiento con XELODA® y, en algunos casos, hasta un mes después de suspender XELODA®. En un estudio de interacción clínica, después de administrar una dosis única de 20 mg de warfarina, el tratamiento con XELODA® incrementó el ABC de la warfarina-S en un 57%, con un incremento del 91% en el valor del rango normalizado internacional (INR). Los pacientes que toman anticoagulantes derivados de la cumarina en forma concomitante con XELODA®, deben ser monitoreados regularmente en alteraciones en los parámetros de coagulación (PT o INR), y si es necesario, ajustar la dosis del anticoagulante. Sustratos 2C9 del citocromo P450: no se han realizado estudios formales de interacción entre capecitabina y fármacos que se sabe metabolizan a la isoenzima 2C9 del citocromo P450. Deberá tenerse cuidado al co-administrar XELODA® con estos fármacos. Fenitoína: se ha reportado aumento de las concentraciones plasmáticas de fenitoína, durante su uso concomitante con XELODA®. No se han realizado estudios formales de interacción farmacológica con la fenitoína, pero se considera que el mecanismo de acción es la inhibición del sistema de la isoenzima CYP2C9 por parte de la capecitabina (ver Anticoagulantes cumarínicos). En los pacientes que toman concomitantemente fenitoína con XELODA®, se deberán monitorear regularmente las concentraciones plasmáticas de fenitoína. Interacción con los alimentos: en todos los estudios clínicos se proporcionó instrucciones a los pacientes para que tomaran XELODA® 30 minutos después de los alimentos. Debido a que los datos actuales de seguridad y eficacia están basados en la administración con alimentos, se recomienda que XELODA® se administre con alimentos. Antiácidos: se investigó el efecto de un antiácido que contiene hidróxido de aluminio e hidróxido de magnesio, sobre la farmacocinética de la capecitabina, en pacientes con cáncer. Hubo un pequeño incremento en las concentraciones plasmáticas de la capecitabina y un metabolito (5'-DFCR); no hubo efecto en los 3 metabolitos principales (5'-DFUR, 5-FU y FBAL). Leucovorina (ácido folínico): se investigó el efecto de la leucovorina sobre la farmacocinética de la capecitabina, en pacientes con cáncer. La leucovorina no tuvo efecto sobre la farmacocinética de la capecitabina ni de sus metabolitos. Sin embargo, la leucovorina tiene un efecto sobre la farmacodinámica de XELODA® y su toxicidad puede aumentarse por la leucovorina. Sorivudina y análogos: se ha descrito en la literatura médica, una interacción farmacológica clínicamente importante entre sorivudina y 5-FU, resultante de la inhibición de la dihidropirimidina deshidrogenasa por la sorivudina. Esta interacción que conduce a un aumento en la toxicidad de la fluoropirimidina, es potencialmente fatal. Por lo tanto, XELODA® no deberá administrarse concomitantemente con sorivudina o sus análogos químicamente relacionados, tal como brivudina (ver Contraindicaciones). Debe haber por lo menos un período de espera de 4 semanas entre el fin de la terapia con brivudina y el inicio de la terapia con XELODA® Oxaliplatino: no se presentaron diferencias clínicamente significativas en la exposición a la capecitabina o sus metabolitos, platino libre o platino total cuando la capecitabina y oxaliplatino se administraban en combinación, con o sin bevacizumab Bevacizumab: no hubo efecto clínicamente significativo del bevacizumab sobre los parámetros farmacocinéticos de capecitabina o sus metabolitos.
Alteraciones en los resultados de pruebas de laboratorio: La siguiente tabla muestra las alteraciones en pruebas de laboratorio observadas en 995 pacientes, independientemente de la relación con XELODA® como tratamiento adyuvante de cáncer colorrectal.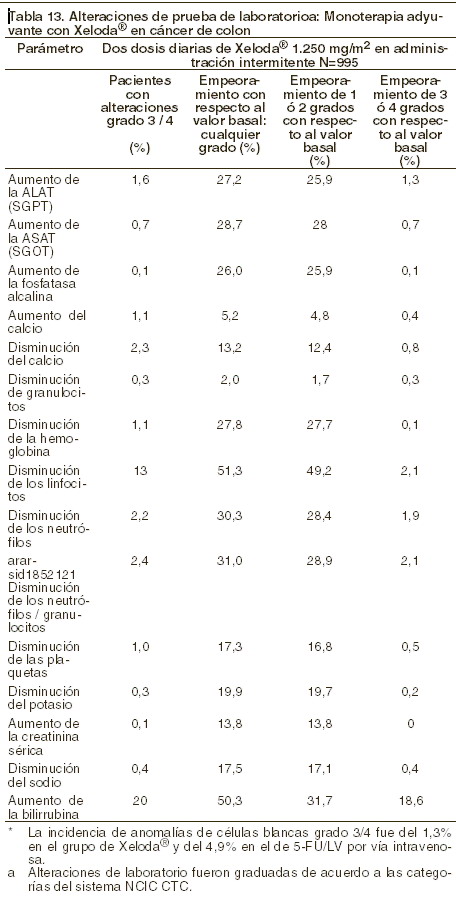
La siguiente tabla muestra las alteraciones de laboratorio observadas en 949 pacientes, sin importar su relación con el tratamiento de XELODA® en cáncer de mama metastásico y cáncer colorrectal.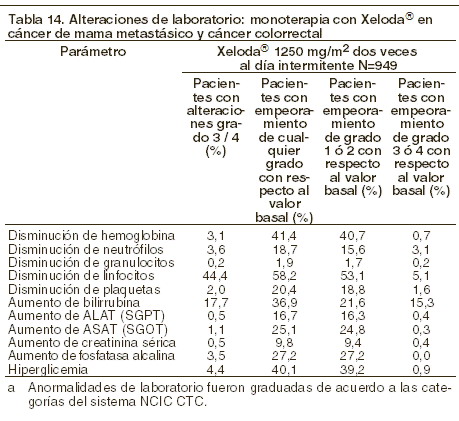
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En los estudios de toxicidad reproductiva en animales, la administración de capecitabina fue teratógena y letal para el embrión. Estos hallazgos son efectos esperados de los derivados de las fluoropirimidinas. La capecitabina debe ser considerada como un teratógeno potencial para el humano. XELODA® no debe utilizarse durante el embarazo.
Dosis y vía de administración: XELODA® Tabletas recubiertas deben ser deglutidas con agua en dos tomas, 30 minutos después de los alimentos (aprox. 200 ml de agua después del desayuno y después de la cena). Monoterapia: cáncer de colon, colorrectal y mama: la dosis inicial recomendada de XELODA®monoterapia es de 1.250 mg/m2, administrada dos veces al día (mañana y noche: equivalente a una dosis total de 2.500 mg/m2 al día), durante 2 semanas, seguido por un período de descanso de siete días. Terapia en combinación: cáncer de mama: en combinación con docetaxel, la dosis inicial recomendada de XELODA® es de 1.250 mg/m2 dos veces al día durante 2 semanas, seguidas de un período de descanso de siete días, combinada con 75 mg/m2 de docetaxel en infusión intravenosa de 1 hora cada 3 semanas. La premedicación, de acuerdo a la información para prescribir de docetaxel, deberá iniciarse antes de la administración en pacientes que reciben la combinación con XELODA®. Cáncer colorrectal y gástrico: en el tratamiento en combinación, la dosis inicial recomendada de XELODA® debe ser reducida de 800 a 1.000 mg/m2, administrada dos veces al día durante 2 semanas, seguido por un período de descanso de 7 días, o 625 mg/m2 dos veces al día diariamente cuando se administre continuamente (ver Eficacia). La inclusión de agentes biológicos en un régimen de combinación no tiene efecto en la dosis inicial de XELODA®. La premedicación para mantener una adecuada hidratación y efecto antiemético, de acuerdo a la información para prescribir de cisplatino y oxaliplatino, deberá iniciarse antes de la administración en pacientes que reciben la combinación con XELODA® + oxaliplatino o cisplatino. La dosis de XELODA® se calcula de acuerdo al área de superficie corporal. Las siguientes tablas muestran ejemplos de los cálculos de dosis estándar y reducida (ver la sección "Ajustes de dosificación durante el tratamiento") para una dosis inicial de XELODA® ya sea de 1.250 mg/m2 o 1.000 mg/m2.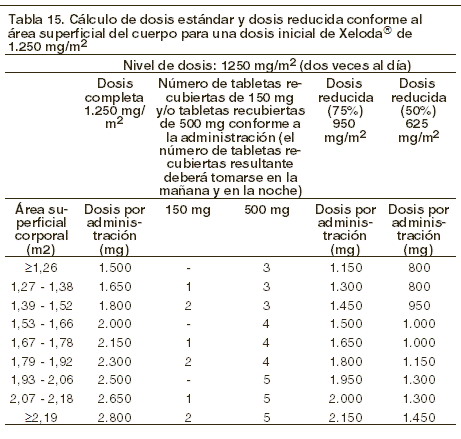
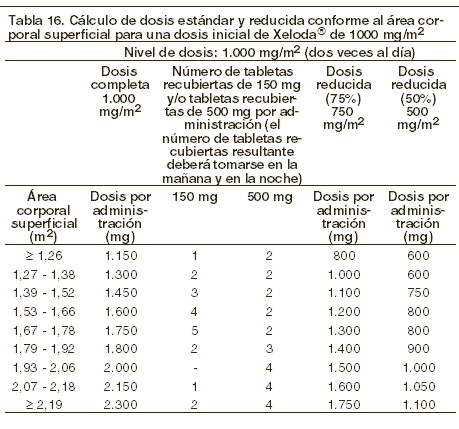
Ajustes de dosificación durante el tratamiento: general: la toxicidad debida a la administración de XELODA® puede ser manejada con tratamiento sintomático y/o modificación de la dosis de XELODA® (interrupción del tratamiento o reducción de la dosis). Una vez que se haya reducido la dosis, no se deberá incrementar en un futuro. Para aquellas toxicidades que el médico tratante considere que no sea probable que se conviertan en serias o que pongan en riesgo la vida del paciente, el tratamiento se puede continuar a la misma dosis sin reducción o interrupción. Las modificaciones de la dosis no son recomendadas en los eventos grado 1. La terapia con XELODA® deberá ser interrumpida si se presentan eventos adversos grado 2 o 3. Una vez que los eventos adversos se hayan resueltos o disminuido su intensidad a grado 1, la terapia con XELODA® podrá ser reiniciada hasta la dosis completa, o ajustada de acuerdo a la tabla 17. Si se presenta un evento adverso grado 4, la terapia deberá ser suspendida o interrumpida hasta que el evento se haya resuelto o disminuido a grado 1, y el tratamiento puede reiniciarse con la mitad de la dosis original. Los pacientes que toman XELODA® deben ser informados de la necesidad de interrumpir el tratamiento inmediatamente si ocurre alguna toxicidad moderada o severa. Las dosis de XELODA® omitidas por toxicidad, no deben reponerse. Hematología: pacientes con cuenta basal de neutrófilos < 1,5 x 109/l y/o cuentas de trombocitos < 100 x 109/l no deben ser tratados con XELODA®. Si las evaluaciones de laboratorio no programadas durante algún ciclo de tratamiento demuestran toxicidad hematológica grado 3 o 4, deberá interrumpirse el tratamiento con XELODA®. La siguiente tabla muestra las modificaciones de dosis recomendadas, seguidas a la toxicidad relacionadas a XELODA®: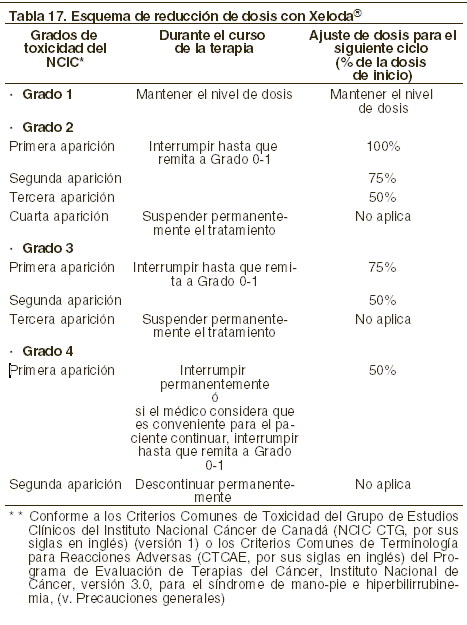
Terapia de combinación general: deben hacerse modificaciones de la dosis por toxicidad cuando XELODA® se usa en combinación con otras terapias conforme a la tabla 17 anterior para XELODA® y conforme a la información de prescripción correspondientes para los otros agentes. Al inicio de un ciclo de tratamiento, si se tiene indicado un retraso en el tratamiento para XELODA® o el otro agente, entonces la administración de todos los agentes debe retrasarse hasta que se cumplan los requerimientos para el reinicio de todos los fármacos. Durante un ciclo de tratamiento para aquellas toxicidades que el médico tratante las consideró como no relacionadas con el XELODA®, XELODA® debe continuarse y ajustarse la dosis del otro agente conforme a la información de prescripción correspondiente. Si se debe discontinuar permanente a los otros agentes, el tratamiento con XELODA® se puede reanudar cuando se cumplan los requerimientos para reiniciar con XELODA®. Esta recomendación aplica a todas las indicaciones y a todas las poblaciones especiales. Instrucciones especiales de dosificación: pacientes con insuficiencia hepática debida a metástasis en hígado: en pacientes con insuficiencia hepática leve a moderada debida a metástasis en hígado, no es necesario el ajuste de dosis inicial. Sin embargo, tales pacientes deben ser vigilados cuidadosamente (ver Farmacocinética en poblaciones especiales e Instrucciones especiales de dosificación). No se han estudiado los pacientes con insuficiencia hepática grave. Pacientes con insuficiencia renal: en pacientes con insuficiencia renal moderada (depuración de creatinina 30-50 ml/min [Cockroft y Gault]) en el registro basal, se recomienda una reducción de 1.250 mg/m2 de la dosis de inicio al 75%. En pacientes con insuficiencia renal leve (depuración de creatinina 51-80 ml/min), no se recomienda el ajuste de la dosis de inicio. Se deberá hacer un monitoreo cuidadoso y la interrupción inmediata del tratamiento, si el paciente desarrolla un evento adverso grado 2, 3 o 4, con ajustes subsecuentes en la dosis, en la tabla 17 (ver Farmacocinética en poblaciones especiales). Si durante el tratamiento el cálculo de la depuración de creatinina disminuye a un valor menor a 30 ml/min, XELODA® debe ser discontinuado. Para cálculos sobre la dosificación, ver tablas 15 y 16. Niños: no se ha establecido la seguridad y eficacia de XELODA® en niños. Ancianos: para la monoterapia de XELODA®, no es necesario ajustar la dosis inicial. Sin embargo, las reacciones adversas severas grado 3 o 4 relacionadas con el tratamiento fueron más frecuentes en pacientes mayores a 80 años de edad en combinación con pacientes más jóvenes. Cuando XELODA® se usó en combinación con otros agentes, los pacientes geriátricos (≥ 65 años de edad) experimentaron más reacciones adversas a medicamento (RAM) grado 3 y 4 y RAF que conllevaron a la discontinuación, respecto de pacientes más jóvenes. Se recomienda el monitoreo cuidadoso de los pacientes ancianos. En combinación con docetaxel: se ha observado un aumento en la incidencia de eventos adversos grado 3 o 4, así como eventos adversos graves, relacionados con el tratamiento, en pacientes de 60 o más años de edad. En los pacientes con edad de 60 años o más, tratados con la combinación de XELODA®/docetaxel, se recomienda una reducción de la dosis de inicio de XELODA® al 75% (950 mg/m2 dos veces al día). Para el cálculo de la dosis, ver la tabla 16. En combinación con irinotecan: para pacientes de 65 años de edad o mayores, se recomienda una reducción de la dosis inicial de XELODA® a 800 mg/m2 dos veces al día.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Las manifestaciones de sobredosis aguda consisten en náusea, vómito, diarrea, mucositis, irritación, sangrado gastrointestinal y depresión de la médula ósea. El manejo médico de la sobredosis deberá consistir en medidas médicas de apoyo y terapéuticas de rutina, dirigidas a corregir las manifestaciones clínicas presentes y prevenir posibles complicaciones.
Presentación(es): Caja con 60 tabletas recubiertas de 150 mg. Caja con 120 tabletas recubiertas de 500 mg.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30 °C y en lugar seco.
Leyendas de protección: Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología, con experiencia en quimioterapia antineoplásica. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo y la lactancia. Literatura exclusiva para médicos.
Nombre y domicilio del laboratorio: Hecho en Estados Unidos por: Hoffmann - La Roche Inc. 340 Kingsland Street, Nutley, New Jersey 07110, EUA y acondicionado y distribuido por: Productos Roche S.A. de C.V. Vía I. Fabela Nte. No. 1536-B, Col. Parque Industrial, 50030, Toluca, México o hecho en México por: Productos Roche S.A. de C.V. Vía I. Fabela Nte. No. 1536-B, Col. Parque Industrial, 50030, Toluca, México y acondicionado por: Syntex S.A. de C.V. Vía I. Fabela Nte. No. 1536-A, Col. Parque Industrial, 50030, Toluca, México. Bajo licencia de: F. Hoffmann - La Roche S.A. Para información adicional sobre este medicamento, comuníquese a nuestro centro de información médica, Tel. (01)(55) 52585099 y 01-800-8218887 o mexico.info@roche.com. ®Marca registrada.
Número de registro del medicamento: 522M98 SSA IV.
Clave de IPPA: BEAR-083501414B0002/RM2009