XOLAIR
NOVARTIS
Denominación genérica: Omalizumab.
Forma farmacéutica y formulación: Solución. El frasco ámpula contiene: Omalizumab 202.5 mg, Excipiente cbp. La ampolleta con diluyente contiene: Agua inyectable 2 mL, Hecha la mezcla el frasco ámpula contiene: Omalizumab 150 mg/1.2 mL. La jeringa prellenada contiene: Omalizumab 75 mg y 150 mg Vehículo c.b.p 0.5 mL y 1 mL Contenido total en la jeringa prellenada 75 mg/0.5 mL y 50 mg/mL
Indicaciones terapéuticas: Asma Alérgica: XOLAIR® (omalizumab) está indicado para el tratamiento de adultos y niños (de 6 años o mayores) afectados de asma alérgica moderada a grave, persistente, cuyos síntomas no logran controlarse a pesar de dosis elevadas de corticoesteroides inhalados.XOLAIR® ha demostrado que disminuye la incidencia de exacerbaciones asmáticas en estos tipos de pacientes. La seguridad y la eficacia de XOLAIR® no se han establecido para otras condiciones alérgicas. Urticaria Crónica Espontánea (UCE): XOLAIR® (omalizumab) está indicado para el tratamiento adyuvante de los adultos y adolescentes (mayores de 12 años) afectados de urticaria crónica espontánea resistente al tratamiento con antihistamínicos.
Farmacocinética y farmacodinamia: Farmacocinética: Características Generales: Absorción: Tras la administración subcutánea, el omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. La farmacocinética de omalizumab es lineal a dosis mayores que 0.5 mg/kg. Se obtienen curvas similares de concentración sérica (de omalizumab) y tiempo cuando se administran las formulaciones liofilizada o líquida de XOLAIR®. Distribución: In vitro, el omalizumab forma complejos de reducido tamaño con la IgE. Tanto in vitro como in vivo no se han observado complejos precipitantes ni complejos con pesos moleculares superiores a 1x106 Daltons. Los estudios de distribución hística en macacos no evidenciaron una captación específica de I125-omalizumab por parte de ningún órgano o tejido. Eliminación: La depuración del omalizumab comprende procesos de depuración de lgG y de depuración a través de uniones específicas y de formación de complejos con su ligando específico, la lgE. La eliminación hepática de lgG incluye una degradación en las células endoteliales y reticuloendoteliales del hígado. La lgG inalterada se elimina asimismo en la bilis. En los estudios con ratones y monos, los complejos de omalizumab: lgE se eliminaron a través de interacciones con los receptores Fc? en el sistema reticuloendotelial del hígado a una velocidad generalmente mayor que la depuración de las lgG. Pacientes con asma alérgica: Absorción: Después de administrar una dosis subcutánea única a pacientes adultos o adolescentes con asma, el omalizumab se absorbe con lentitud y alcanza su mayor concentración sérica al cabo de 7 u 8 días, en promedio. Tras la administración de dosis repetidas de omalizumab, el área bajo la curva de concentración sérica y tiempo desde el día 0 al 14 en el estado estacionario llegó a ser hasta seis veces mayor que la obtenida después de administrar la primera dosis. Distribución: Luego de la administración subcutánea, el volumen aparente de distribución de omalizumab en los pacientes asmáticos es de 78 ± 32 mL/kg. Eliminación: En los pacientes asmáticos, la vida media de eliminación sérica del omalizumab promedia los 26 días y su depuración aparente es de 2.4 ± 1.1 ml/kg/día, en promedio. La duplicación del peso corporal hace que la depuración aparente sea aproximadamente el doble. Edad, raza o grupo étnico, género e índice de masa corporal: Se analizó la farmacocinética poblacional del omalizumab para determinar la influencia de las características demográficas. Los análisis de estos datos indican que no es necesario ajustar la dosis en los pacientes asmáticos en función de la edad (6-76 años), la raza, el grupo étnico, el género o el índice de masa corporal. Pacientes con urticaria crónica espontánea (UCE): Absorción: Tras la administración de una dosis subcutánea única a pacientes adultos o adolescentes con UCE, el omalizumab se absorbió con lentitud y alcanzó su mayor concentración sérica al cabo de 6 u 8 días, en promedio. En pacientes con UCE, el omalizumab presenta una farmacocinética lineal en la gama de dosis de 75 a 600 mg administradas una sola vez por vía subcutánea. Luego de la administración de dosis de 75, 150 o 300 mg cada 4 semanas, la concentración sérica mínima de omalizumab aumenta de forma proporcional a la dosis. Distribución: Según un estudio de farmacocinética poblacional, la distribución de omalizumab en los pacientes con UCE es similar a la de los pacientes con asma alérgica. Eliminación: Las simulaciones farmacocinéticas poblacionales indicaron que, en los pacientes con UCE, la vida media de eliminación sérica del omalizumab promedia los 24 días en el estado estacionario y la depuración aparente en el estado estacionario, los 240 mL/día (que corresponde a 3.0 mL/kg/día en un paciente de 80 kg). Edad, raza o grupo étnico, género, peso corporal, índice de masa corporal, lgE basal, autoanticuerpos anti-Fc?RI, comedicación: Los efectos de las covariables demográficas y de otros factores sobre la exposición al omalizumab se evaluaron mediante análisis de farmacocinética poblacional. También se evaluaron los efectos de covariables analizando la relación que existía entre las distintas concentraciones de omalizumab y las respuestas clínicas. Tales análisis han revelado que no hace falta ajustar la dosis en los pacientes con UCE en función de la edad (12 a 75 años), la raza o el grupo étnico, el género, el peso corporal, el índice de masa corporal, la lgE basal, los autoanticuerpos anti-Fc?RI o el uso simultáneo de antihistamínicos H² o de antagonistas de los receptores de leucotrienos (ARL). Insuficiencia renal o hepática: No se dispone de datos farmacocinéticos ni farmacodinámicos en pacientes con disfunción renal o hepática aquejados de asma alérgica o UCE (ver Precauciones generales). Farmacodinamia: Código ATC: Grupo farmacoterapéutico: Otros agentes contra padecimientos obstructivos de las vías respiratorias. ATC: R03DX05. Características Generales: Omalizumab es un anticuerpo monoclonal humanizado, obtenido por ingeniería genética, que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 (kappa) que contiene, enmarcadas por regiones humanas, las regiones determinantes de complementariedad de un anticuerpo murino que se une a la IgE. Pacientes con asma alérgica: La cascada alérgica se inicia cuando el alergeno interconecta las lgE que se fijan a los receptores Fc?RI (receptores con gran afinidad por la lgE) en la superficie de los mastocitos y los basófilos. Ello produce la desgranulación de esas células efectoras y la liberación de histaminas, leucotrienos, citocinas y otros mediadores. Dichos mediadores guardan una relación causal con la fisiopatología del asma alérgica, que incluye la formación de un edema en las vías respiratorias, la contracción del músculo liso y una alteración de la actividad celular asociada al proceso inflamatorio. También contribuyen a producir los signos y síntomas de la enfermedad alérgica, a saber, broncoconstricción, producción de moco, sibilancias, disnea, opresión torácica, congestión nasal, estornudos, picazón nasal, rinorrea, picazón ocular y ojos llorosos. El omalizumab se fija a la lgE e impide que ésta se una al receptor Fc?RI, reduciendo así la cantidad de lgE libre disponible para iniciar la cascada alérgica. El tratamiento con omalizumab de sujetos atópicos causó un pronunciado descenso del número de receptores Fc?RI. Además, la liberación in vitro de histamina de los basófilos aislados de individuos que habían recibido XOLAIR® disminuyó cerca del 90% tras la estimulación con un alergeno, en comparación con los valores previos al tratamiento. En los pacientes asmáticos de los estudios clínicos, las concentraciones séricas de lgE libre disminuyeron de forma dependiente de la dosis en la hora siguiente a la administración de la primera dosis y se mantuvieron constantes entre dos administraciones. La reducción media de las concentraciones séricas de lgE libre fue superior al 96% cuando se utilizaron las dosis recomendadas. Las concentraciones séricas de lgE total (es decir, unida y no unida a proteínas) aumentaron tras la primera dosis debido a la formación de complejos de omalizumab: lgE que se eliminan más lentamente que la lgE libre. Dieciséis semanas después de la primera dosis, las concentraciones séricas de lgE total eran, en promedio, unas cinco veces mayores que las cifras anteriores al tratamiento cuando se usaron estudios convencionales. Tras interrumpir la administración de XOLAIR®, el aumento de la concentración de lgE total y la disminución de la concentracíón de lgE libre inducidos por XOLAIR® revirtieron sin que se observase ningún efecto de rebote en las concentraciones de lgE después del periodo de reposo farmacológico. Las concentraciones de lgE total no regresaron a sus niveles preterapéúticos sino hasta un año después de la retirada de XOLAIR®. Pacientes con urticaria crónica espontánea (UCE): Existen varias teorías sobre la etiología de la UCE; una de ellas apunta a un origen autoinmunitario de la enfermedad. Se han aislado anticuerpos autoinmunitarios contra la lgE y su receptor, Fc?RI, del suero de algunos pacientes que padecían UCE. Tales autoanticuerpos pueden activar los basófilos o mastocitos y provocar la liberación de histamina. Una de las hipótesis del modo de acción del omalizumab en la UCE es que reduce las concentraciones de lgE libre en la sangre y posteriormente en la piel. Ello causa un descenso del número de receptores superficiales de la lgE y de ese modo se reduce la consiguiente transcripción de señales a través de la vía del Fc?RI, lo cual redunda en inhibición de las reacciones inflamatorias y de activación celular. Como corolario, disminuyen la frecuencia y la intensidad de los síntomas de UCE. Otra hipótesis es que la disminución de la cantidad circulante de lgE libre produce una desensibilización rápida e inespecífica de los mastocitos cutáneos. El descenso del número de Fc?RI puede contribuir a mantener la respuesta. En los estudios clínicos de pacientes con UCE, el tratamiento con omalizumab produjo una reducción, dependiente de la dosis, de la cantidad de lgE libre y un aumento de la concentración de lgE total en el suero, de forma similar a lo que se observa en los pacientes con asma alérgica. La máxima disminución de lgE libre se observó 3 días después de la primera dosis subcutánea. Tras la administración repetida del medicamento cada 4 semanas, las cifras séricas de lgE libre anteriores a cada administración permanecieron estables entre las semanas 12 y 24 de tratamiento. Las cifras séricas de lgE total aumentaron después de la primera dosis debido a la formación de complejos omalizumab: lgE de eliminación más lenta que la lgE libre. Al cabo de 12 semanas de administración repetida del medicamento en dosis de entre 75 y 300 mg cada 4 semanas, las cifras séricas medias de lgE anteriores a la administración eran dos o tres veces mayores que las cifras determinadas antes de la terapia (preterapéuticas) y permanecieron estables entre las semanas 12 y 24 de tratamiento. Tras la retirada de XOLAIR®, durante un periodo de observación sin tratamiento de 16 semanas, las cifras de lgE libre aumentaron y las de lgE total disminuyeron hasta acercarse a los niveles preterapéuticos.
Contraindicaciones: Hipersensibilidad al principio activo o a cualquiera de los excipientes, embarazo y lactancia.
Restricciones de uso durante el embarazo y la lactancia: Mujeres en edad fértil: No existen recomendaciones especiales para las mujeres en edad fértil. Embarazo: El uso durante el embarazo está contraindicado.No se han realizado estudios adecuados ni apropiadamente controlados con el omalizumab en las mujeres embarazadas. Las IgG atraviesan la barrera placentaria. Dado que los estudios de la reproducción animal no siempre permiten predecir la respuesta en humanos, XOLAIR® no debe administrarse durante el embarazo, salvo cuando sea estrictamente necesario. Se han estudiado los efectos del omalizumab en la reproducción de macacos de la especie Macaca fascicularis (macaco de Java). Las dosis subcutáneas de hasta 75 mg de omalizumab/kg semanalmente (al menos 8 veces la dosis clínica máxima recomendada en mg/kg por un periodo de 4 semanas) no indujeron toxicidad materna o embrionaria ni teratogenia cuando se administraron durante la organogénesis, ni tampoco efectos adversos sobre el crecimiento fetal o neonatal cuando se administraron durante el último periodo de gestación, el alumbramiento o la lactancia. Aunque no se han observado efectos clínicamente significativos en las plaquetas de los pacientes, las dosis de omalizumab superiores a las clínicamente aprobadas se han asociado con reducciones dependientes de la edad de las plaquetas sanguíneas en primates, en las cuales los animales más jóvenes se vieron relativamente más afectados. En estudios reproductivos efectuados en macacos de Java no se apreciaron signos clínicos de trombocitopenia en los neonatos nacidos de madres que habían recibido dosis de hasta 75 mg de omalizumab/kg; no obstante, no se determinaron los recuentos de plaquetas en estas crías. Lactancia: Aunque no se ha investigado la presencia de omalizumab en la leche materna humana, las IgG son excretadas por esta vía y, por consiguiente, cabe esperar que omalizumab pase a la leche materna humana. Se desconocen la capacidad de absorción del omalizumab y de daño al lactante; se debe tener cuidado cuando se administre XOLAIR® a una madre lactante, el médico y la paciente deberán decidir si se suspende la lactancia o se suspende el tratamiento. Se valoró la excreción láctea del omalizumab en hembras de macacos de Java que habían recibido dosis subcutáneas de 75 mg/kg/semana. Las concentraciones séricas de omalizumab en el neonato tras una exposición in utero y de 28 días en el periodo de lactancia variaban entre un 11 y un 94% de la concentración sérica materna. Las concentraciones lácteas de omalizumab eran un 0.15% de la concentración sérica materna. Fertilidad: No existen datos de alteración de la fertilidad en adultos para omalizumab. En los estudios preclínicos diseñados específicamente realizados en macacos de Java, incluyendo estudios de apareamiento, no se observó alteración alguna en la fertilidad de las hembras o los machos después de administrar en dosis repetidas de omalizumab de hasta 75 mg/kg.
Reacciones secundarias y adversas: Asma Alérgica:Resumen del perfil de seguridad: Las reacciones adversas más frecuentes en los estudios clínicos con adultos y con pacientes adolescentes de más de 12 años de edad fueron cefaleas y reacciones relacionadas con la inyección en sí, e incluían dolor, hinchazón, eritema, y prurito. En estudios clínicos en pacientes de 6 a < 12 años de edad las reacciones más comúnmente reportadas fueron dolor de cabeza, fiebre y dolor abdominal alto. La mayoría de los eventos fueron leves o moderados en severidad. Resumen estratificado de las reacciones adversas de los estudios clínicos: La Tabla 1 recoge las reacciones adversas registradas en los estudios clínicos en la población total con asma alérgica tratada con XOLAIR® del análisis de la seguridad, desglosadas por clase de órgano, aparato o sistema y por frecuencia. Según su frecuencia se clasifican en: Muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); infrecuentes (≥ 1/1,000 a < 1/100); raras (≥ 1/10,000 a < 1/1,000); muy raras ( < 1/10,000).
Tabla 1. Reacciones adversas registradas en los estudios clínicos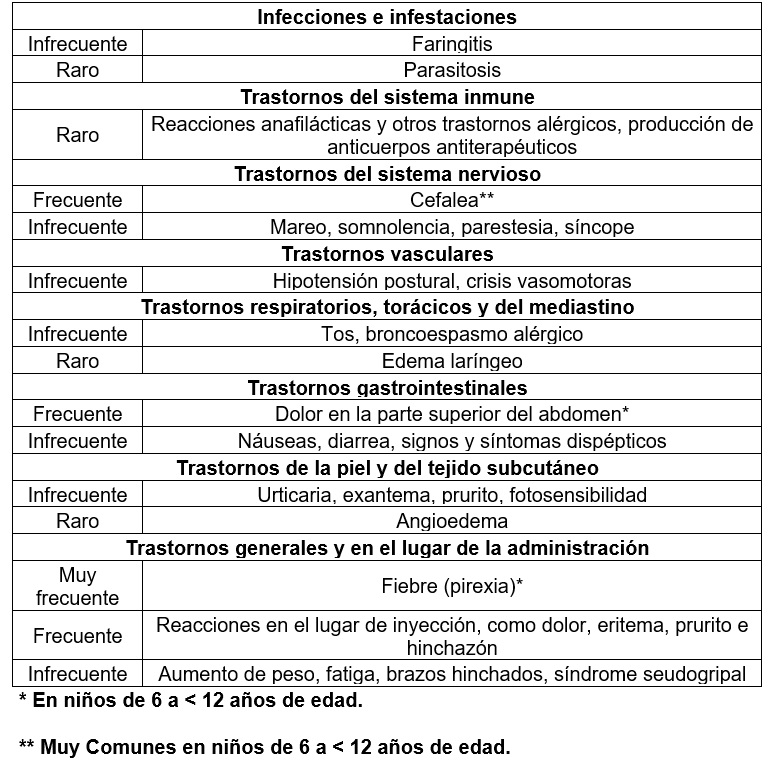
Las frecuencias de reacciones adversas en los pacientes del grupo que recibió el tratamiento activo fueron muy similares a las del grupo de control. Lista de reacciones adversas mencionadas en notificaciones espontáneas desde la comercialización del producto: Se han identificado las siguientes reacciones a través de reportes espontáneos. Trastornos del sistema inmune (ver Precauciones generales): Se han reportado anafilaxia y reacciones anafilactoides posteriormente a la primer o subsecuentes administraciones, enfermedad del suero. Trastornos de la Piel y subcutáneos: Alopecia. Trastornos de la sangre y tejido linfático: Trombocitopenia idiopática severa. Trastornos respiratorios, torácicos y del mediastino: Vasculitis granulomatosa alérgica (Síndrome Churg Strauss)Trastornos músculo esqueléticos y del tejido conectivo: Artralgia, mialgia, inflamación articular. Urticaria crónica espontánea (UCE): Resumen del perfil toxicológico: La seguridad y la tolerabilidad del omalizumab se evaluaron en 975 pacientes con UCE, de los cuales 733 recibieron omalizumab durante 12 semanas en dosis de 75, 150 o 300 mg cada cuatro semanas, y 242, el placebo. En 490 de los pacientes tratados con omalizumab la administración duró 24 semanas. De los pacientes tratados con omalizumab durante 12 semanas, 175 recibieron la dosis recomendada de 150 mg, y 412, la de 300 mg. De los pacientes tratados con omalizumab durante 24 semanas, 87 recibieron la dosis recomendada de 150 mg, y 333, la de 300 mg. Durante los estudios clínicos en adultos y adolescentes (mayores de 12 años de edad), las reacciones adversas más frecuentemente observadas fueron cefalea y nasofaringitis. Resumen tabulado de las reacciones adversas observadas en los estudios médicos con las dosis recomendadas (150 o 300 mg): La Tabla 2 presenta, ordenadas por clase de órgano, aparato o sistema del MedDRA, las reacciones adversas descritas con las dosis recomendadas (150 mg y 300 mg) en los tres estudios de fase III. Tales reacciones se observaron en ≥ 1% de los pacientes de cualquier grupo de tratamiento, con mayor frecuencia (≥ 2%) en los que recibieron el omalizumab que en los tratados con el placebo, según una evaluación médica. Dentro de cada clase de órgano, aparato o sistema las reacciones adversas figuran por orden decreciente de frecuencia. La categoría de frecuencia de cada reacción adversa se define según la siguiente convención (CIOMS III): muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); infrecuente (≥ 1/1000 a < 1/100); rara (≥ 1 /10 000 a < 1/1000); muy rara ( < 1/10 000); (de frecuencia) desconocida (no se puede calcular con los datos disponibles).
Tabla 2. Reacciones adversas registradas en la base de datos conjuntos de seguridaden la UCE (desde el día 1 hasta la semana 12) con las dosis recomendadas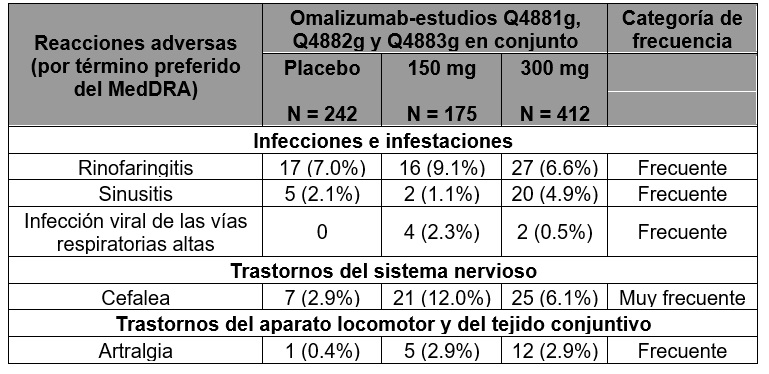
Acontecimientos descritos adicionalmente en cualquier momento del periodo de tratamiento entre el día 1 y la semana 24 (estudios Q4881g y Q4883g) que cumplían los criterios de reacciones adversas: Infecciones e infestaciones: Infección de las vías respiratorias altas (placebo: 3.1%, 150 mg: 3.4%, 300 mg: 5.7%), infección de las vías urinarias (placebo: 1.8%, 150 mg: 4.6%, 300 mg: 2.4%). Trastornos del sistema nervioso: Cefalea sinusal [por congestión de los senos paranasales] (placebo: 0%, 150 mg: 2.3%, 300 mg: 0.3%). Trastornos del aparato locomotor y del tejido conjuntivo: Mialgia (placebo: 0%, 150 mg: 2.3%, 300 mg: 0.9%), dolor en las extremidades (placebo: 0%, 150 mg: 3.4%, 300 mg: 0.9%), dolor osteomuscular (placebo: 0%, 150 mg: 2.3%, 300 mg: 0.9%). Trastornos generales y en el lugar de la administración: Pirexia (placebo: 1.2%, 150 mg: 3.4%, 300 mg: 0.9%). Reacciones en el lugar de la inyección: Durante los estudios, las reacciones en el lugar de la inyección fueron más frecuentes en los pacientes del grupo del omalizumab que en los del placebo (300 mg: 2.7%, 150 mg: 0.6%, placebo: 0.8%) y consistían en inflamación, eritema, dolor, equimosis, prurito, hemorragia y urticaria. Descripción de los aspectos toxicológicos más importantes para las indicaciones de asma alérgica y UCE: Los estudios clínicos en la UCE no han arrojado ningún dato importante que pudiera exigir una modificación de los apartados siguientes. Anafilaxia: En informes posteriores a la comercialización, la frecuencia de la anafilaxia en pacientes expuestos al uso de XOLAIR® se estimó en 0.2% en base a un número total de reacciones anafilacticas observadas desde una exposición estimada de más de 500,000 años paciente. Un historial médico de anafilaxia no relacionado con omalizumab pueden ser un factor de riesgo de anafilaxia después de la administración de XOLAIR®. Neoplasias malignas: En los estudios clínicos iniciales realizados en pacientes (mayores de 12 años de edad) se observó un desequilibrio numérico de casos de cáncer entre el grupo del tratamiento activo y el grupo de referencia. Los casos observados eran infrecuentes ( < 1/100) en ambos grupos de tratamiento activo y de referencia. En un estudio Fase IV de observación subsecuente donde se compararon 5,007 pacientes tratados con XOLAIR® y 2,829 que no fueron tratados con XOLAIR® a los cuales se les dio seguimiento durante un máximo de 5 años, las tasas de incidencia de tumores malignos primarios por cada 1,000 pacientes-año fueron 16.01 para el grupo con XOLAIR® (295/18,426 pacientes-año) y 19.07 en grupo control (190/9,963 pacientes año), respectivamente, lo que no indica un aumento de riesgo de malignidad (cociente de tasas de 0.84, intervalo de confianza del 95%, de 0.62 a 1.13). En un nuevo análisis de estudios clínicos aleatorizados, a doble ciego, controlados con placebo, los cuales incluyeron 4,254 pacientes tratados con XOLAIR® y 3,178 pacientes tratados con placebo, el tratamiento con XOLAIR® no se asoció con un riesgo mayor de malignidad basado en las tasas de incidencia por cada 1,000 pacientes-año de 4.14 (14/3,382 pacientes-año) para los pacientes tratados con XOLAIR® y 4.45 (11/2,474 pacientes-año) en los pacientes tratados con placebo (cociente de tasas 0.93, intervalo de confianza del 95%, de 0.39 a 2.27). El porcentaje de incidencia general de malignidad observado en el programa de estudios clínicos de XOLAIR® fue comparable al registrado en la población general. No hubo casos de neoplasia maligna en los pacientes de entre 6 y < 12 años de edad del grupo del omalizumab de los estudios clínicos y se registró un solo caso de neoplasia maligna en el grupo de referencia. Eventos arteriales tromboembólicos (EAT): En estudios clínicos controlados y durante los análisis interinos de estudio observacional, se observó un desbalance numérico en EAT"s. Los EAT"s incluyeron EVC, ataque isquémico transitorio, infarto del miocardio, angina inestable y muerte de origen cardiovascular (incluyendo muerte de causa desconocida). En el análisis final del estudio observacional, la tasa de EAT por 1,000 pacientes-año fue de 7.52 (115/15,286 pacientes años) para los pacientes tratados con XOLAIR® y 5.12 (51/9,963 pacientes-año) en los del grupo control. En un análisis de variables múltiples controlando por factores riesgo cardiovascular basales disponibles, el riesgo relativo fue de 1.32 (intervalo de confianza del 95%, de 0.91 a 1.91). En un análisis separado de estudios clínicos agrupados que incluyeron todos los estudios clínicos aleatorios, a doble ciego controlados con placebo de 8 o más semanas de duración, la tasa de ETA por 1,000 pacientes-año fue de 2.69 (5/1,856 pacientes-año) para los pacientes tratados con XOLAIR® y 2.38 (4/1,680 pacientes año) en los pacientes tratados con placebo (cociente de tasas 1.13, intervalo de confianza del 95%, de 0.24 a 5.71). Plaquetas: En los estudios clínicos, pocos pacientes presentaron recuentos de plaquetas por debajo del límite inferior del rango normal de laboratorio. Ninguno de estos cambios se asoció con episodios hemorrágicos o con una disminución de la hemoglobina. No se reportó ningún patrón de disminución sistemática en el conteo de plaquetas en humanos (pacientes mayores de 6 años de edad) a diferencia de lo reportado en otros primates (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Parasitosis: En los pacientes alérgicos expuestos crónicamente a un riesgo elevado de padecer helmintiasis, un estudio controlado con placebo reveló un ligero incremento numérico en la tasa de infección algo superior con omalizumab, aunque este incremento no fue estadísticamente significativo. No variaron el curso, la gravedad ni la respuesta al tratamiento de las infecciones (Ver Precauciones generales).
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: No hubo evidencias de reacciones anafilácticas sistémicas por degranulación de mastocitos en los macacos de Java adultos y jóvenes. En todos los estudios con primates se hallaron complejos circulantes IgE: omalizumab, pero tras la administración de omalizumab no se encontraron signos de enfermedad mediada por inmunocomplejos en órgano alguno (incluido el riñón). Los complejos de IgE de omalizumab no fijan complemento ni median la citotoxicidad dependiente de complemento.La administración crónica de omalizumab en dosis de hasta 250 mg/kg (lo cual es al menos 14 veces la dosis terapéutica más alta recomendada en mg/kg, como indica la tabla de dosificación) fue bien tolerada en los primates jóvenes y adultos, salvo la reducción del recuento de plaquetas, dependiente de la dosis, observada en diversas especies de primates con concentraciones plasmáticas que por lo general superaban la exposición humana máxima de los estudios clínicos cruciales («principales > > ). Los macacos jóvenes fueron más sensibles al efecto a las plaquetas que los adultos. Además, los macacos de Java mostraban signos de inflamación y hemorragia aguda en el sitio de la inyección, congruente con una respuesta inmunitaria local a la administración subcutánea repetida de una proteína heteróloga. No se han realizado estudios oficiales del poder cancerígeno de omalizumab. Se han detectado anticuerpos anti-omalizumab en algunos monos tras la administración subcutánea o intravenosa. Cabe esperar que ello suceda al administrar una proteína heteróloga. Algunos animales no pudieron ser evaluados debido a las concentraciones plásmaticas elevadas de omalizumab, a las concentraciones elevadas de lgE o a ambas cosas a la vez. Sin embargo, las elevadas concentraciones plasmáticas de omalizumab se mantuvieron durante el periodo de tratamiento de los estudios, y no hubo signos evidentes de toxicidad por la presencia de anticuerpos antiomalizumab. Los estudios reproductivos, de excreción en leche materna y de fertilidad en animales, están descritos en la sección Restricciones de uso durante el embarazo y la lactancia.
Interacciones medicamentosas y de otro género: Las enzimas del citocromo P450, las bombas de expulsión y los mecanismos de unión a proteínas no participan en la depuración del omalizumab; por consiguiente, la probabilidad de que ocurran interacciones farmacológicas es reducida. No se han realizado estudios de interacción medicamentosa o vacunas propiamente dichos con XOLAIR®. No hay motivos farmacológicos para esperar interacciones entre los medicamentos comúnmente prescritos contra el asma o la UCE y el omalizumab. Asma alérgica: En los estudios clínicos, XOLAIR® se utilizó normalmente asociado a corticoesteroides inhalados y orales, beta-agonistas inhalados de acción corta o larga, modificadores de leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que esos medicamentos usuales contra el asma afectasen a la inocuidad de XOLAIR®. Se dispone de escasos datos sobre la administración de XOLAIR® asociada a una inmunoterapia específica (como es la terapia de hiposensibilización). Urticaria crónica espontánea (UCE): En los estudios clínicos de UCE, XOLAIR® se administró junto con antihistamínicos (H1 o H2) y antagonistas de los receptores de leucotrienos (ARL). En los estudios de fase III Q4881g y Q4882g todos los pacientes recibieron antihistamínicos H1, además de XOLAIR® o placebo. En el estudio de fase III Q4883g, todos los pacientes recibieron uno o más de un antihistamínicos H1, y/o antihistamínicos H2 y/o antagonistas de los receptores de leucotrienos además de XOLAIR® o placebo. No hubo indicios de que el perfil toxicológico del omalizumab administrado con esos medicamentos fuera distinto del que se le conoce en el asma alérgica. Además, un análisis farmacocinético poblacional no reveló ningún efecto importante de los antihistamínicos H2 y los antagonistas de los recepores de leucotrienos en la farmacocinética del omalizumab (ver Farmacocinética y farmacodinamia). Uso de XOLAIR® en combinación con terapias inmunosupresoras no se ha estudiado.
Alteraciones en los resultados de pruebas de laboratorio: No se han reportado hasta la fecha.
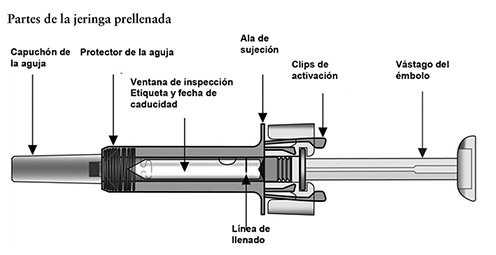
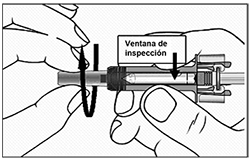
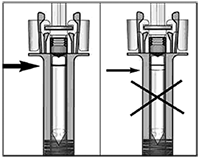
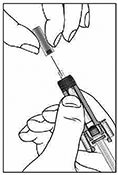
Precauciones generales: Reacciones alérgicas: Al igual que sucede con cualquier proteína, pueden ocurrir reacciones alérgicas locales o sistémicas al administrar omalizumab, incluyendo anafilaxia. Por consiguiente, se debe tener a la mano algún medicamento para el tratamiento inmediato de las reacciones anafilácticas tras la administración de XOLAIR®. El paciente debe saber que estas reacciones son posibles y que si se producen debe acudir al médico de inmediato. Rara vez se han observado reacciones anafilácticas en los estudios clínicos (ver Reacciones secundarias y adversas). En la experiencia posterior a la comercialización se han reportado reacciones anafilácticas y anafilactoides posteriores a la primera o posteriores administraciones de XOLAIR®. Aunque la mayoría de estas reacciones ocurrieron dentro de las primeras 2 horas, algunas ocurrieron después de estas 2 horas. Al igual que otros anticuerpos monoclonales humanizados obtenidos por ingeniería genética, en contadas ocasiones los pacientes pueden generar anticuerpos contra el omalizumab (ver Reacciones secundarias y adversas). La enfermedad del suero y reacciones tipo enfermedad del suero, que son reacciones alérgicas tardías de tipo III, se han visto raramente en pacientes tratados con anticuerpos monoclonales humanizados incluyendo omalizumab. El inicio se ha dado típicamente 1-5 días después de la administración de la primera o subsecuentes administraciones y también después de un tratamiento de larga duración. Los síntomas sugestivos de enfermedad del suero incluyen artritis/artralgia, rash (urticaria u otras formas), fiebre y linfandenopatía. Los antihistamínicos y corticoesteroides pueden ser útiles para prevenir o tratar esta enfermedad y se debe aconsejar a los pacientes el reportar cualquier síntoma sospechoso. Infecciones parasitarias: La IgE puede estar involucrada en la respuesta inmunológica de algunas infecciones. Un estudio comparativo con placebo efectuado en pacientes alérgicos con riesgo crónico elevado de helmintosis reveló un leve aumento de la tasa de infestación con el omalizumab, pero el curso, la gravedad y la respuesta al tratamiento de la infestación no presentaban diferencias. La tasa de infección por helmintos en el programa clínico global, que no fue diseñado para detectar tales infecciones, fue menor de 1 en 1,000 pacientes. Sin embargo, debe tenerse precaución en pacientes con alto riesgo de infección por helmintos, en particular cuando se viaja a áreas endémicas de infecciones por helmintos. En caso de que los pacientes no presenten respuesta al tratamiento anti-helmíntico recomendado, deberá considerarse la descontinuación del tratamiento con XOLAIR®. Recomendaciones Generales: XOLAIR® no está indicado para el tratamiento de las exacerbaciones asmáticas, de los bronco-espasmos o de los estados asmáticos de carácter agudo. No se han investigado los efectos de XOLAIR® en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni tampoco en la prevención de reacciones anafilácticas. No se han investigado suficientemente los efectos de XOLAIR® en dermatitis atópica, rinitis alérgica o alergia alimentaria. Tampoco se conocen los efectos del medicamento en pacientes con enfermedades autoinmunitarias, procesos mediados por inmunocomplejos o insuficiencia renal o hepática pre-existente. Se debe ejercer precaución cuando se administre XOLAIR® a estos pacientes. No se recomienda la descontinuación abrupta de corticoesteroides inhalados o sistémicos después de iniciar la terapia con XOLAIR®. La disminución de corticoesteroides debe realizarse bajo supervisión directa de un médico y pueden requerir realizarse de manera gradual. Aviso para el encargado de aplicar el medicamento: Administrar únicamente por vía subcutánea, no administrar por vía intravenosa o intramuscular. Jeringa prellenada, individuos sensibles al látex: El capuchón desmontable de la aguja de la jeringa prellenada con la solución inyectable de XOLAIR® contiene un derivado del látex o goma natural. Aunque no se detectan restos de látex (goma natural) en el capuchón desmontable de la aguja, todavía no se ha estudiado la seguridad del uso de la solución inyectable de XOLAIR® en jeringa prellenada en personas sensibles al látex.
Dosis y vía de administración: Este producto debe administrarse únicamente por vía subcutánea. No debe administrarse por vía intravenosa ni intramuscular. Dosificación en asma alérgica: La dosis apropiada (mg) y la frecuencia de administración de XOLAIR® se eligen de acuerdo a la concentración basal de la inmunoglobulina E (IgE) (UI/mL), medida antes de iniciar el tratamiento, y al peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración inicial se debe valorar la concentración de IgE en los pacientes mediante una prueba comercial de IgE plasmática total. Basado en estas determinaciones, tal vez sea necesario administrar entre 75 y 600 mg de XOLAIR® por dosis en 1 o 4 inyecciones (en diferente sitio de aplicación). Véase la tabla 3 y la tabla 4 como tablas de conversión y las tablas 5 y 6 para la determinación de la dosis en niños (de 6 a 12 años) y para adolescentes y adultos (mayores de 12 años). Para las dosis de 225,375 ó 525 mg de XOLAIR®, se puede combinar el uso de las jeringas prellenadas de XOLAIR® de 75 mg y 150 mg. Los pacientes que están fuera de los niveles de IgE basales y del peso corporal indicado en la tabla de dosificación no deberían recibir XOLAIR®.
Tabla 3. Conversión de dosis a número de viales, número de inyecciones y volumen total por cada administración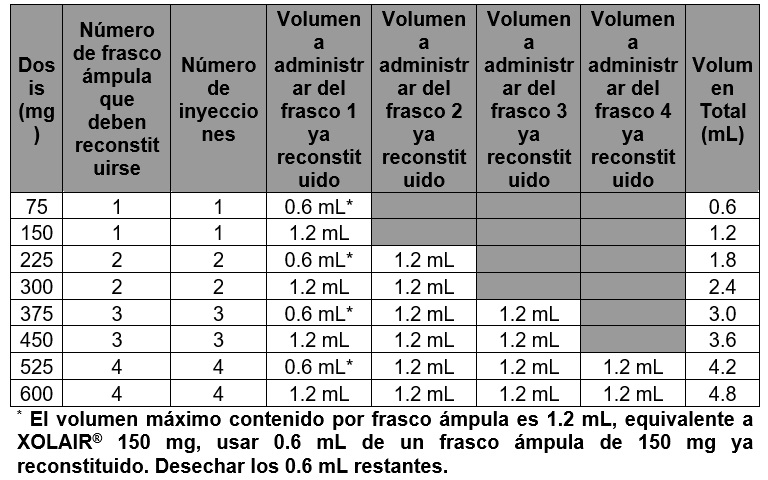
Tabla 4. Conversión de dosis a número de jeringas prellenadas, número de inyecciones y volumen total inyectado en cada administración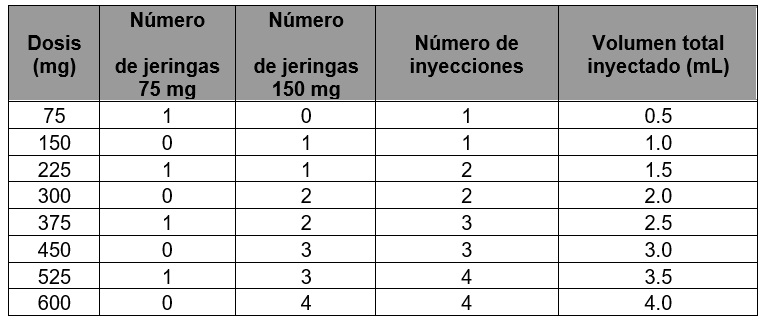
Duración del tratamiento, monitoreo y ajustes de dosis: En los estudios clínicos se observó una reducción en las exacerbaciones asmáticas y en el uso de medicación de rescate, así como mejoría en la calificación de los síntomas durante las primeras 16 semanas de tratamiento. Son necesarias 12 semanas como mínimo para estimar adecuadamente si el paciente responde a XOLAIR®. El uso de XOLAIR® está previsto para largo plazo. La interrupción del tratamiento generalmente resulta en un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son elevados durante el tratamiento y permanecen así hasta un año después de la interrupción del mismo. Por consiguiente, no se recomienda volver a medir las concentraciones de IgE durante el tratamiento con XOLAIR® para determinar la dosis. La determinación de la dosis tras interrupciones de tratamiento de menos de un año de duración debe basarse en las concentraciones plasmáticas de IgE obtenidas al principio. Si el tratamiento con XOLAIR® se ha interrumpido por más de un año, se pueden volver a medir las concentraciones plasmáticas de IgE total a efecto de determinar la dosis. La dosis suele ajustarse por cambios significativos por el peso corporal (véase las tablas 5 y 6).
Tabla 5. Administración cada 4 semanas. Dosis de XOLAIR® (miligramos por dosis) administrada por vía subcutánea cada 4 semanas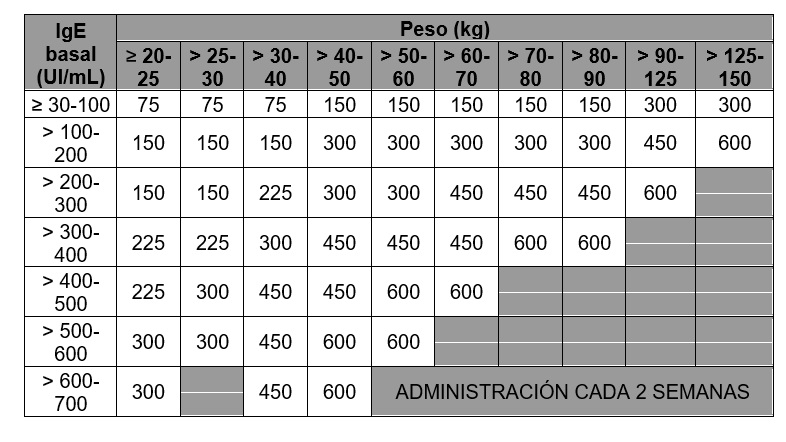
Tabla 6. Administración cada 2 semanas. Dosis de XOLAIR® (miligramos por dosis) administrada por vía subcutánea, cada 2 semanas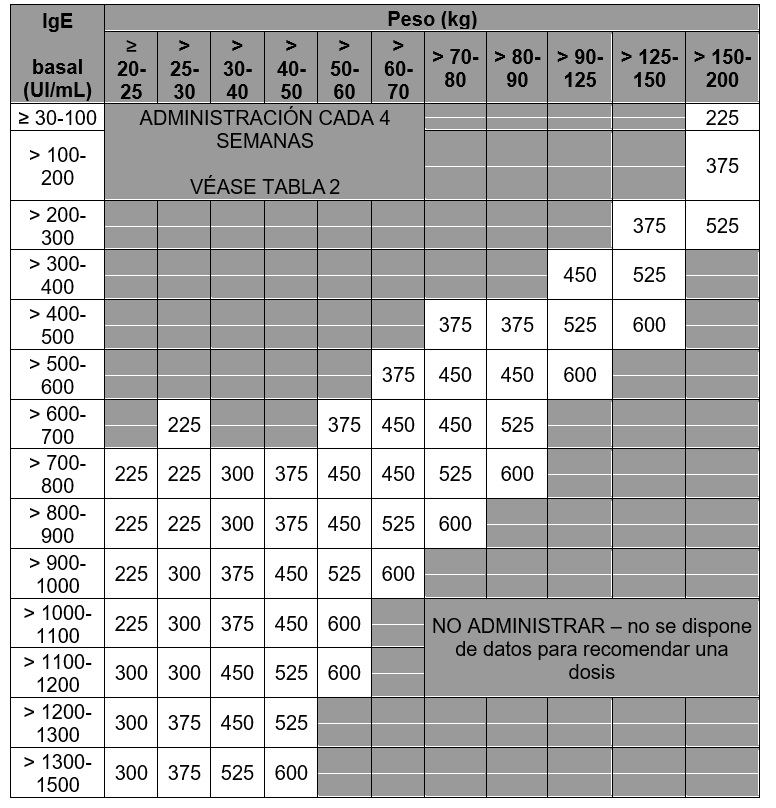
Dosificación en la urticaria crónica espontánea (UCE): La dosis recomendada es de 300 mg, que se administra cada cuatro semanas por inyección subcutánea. Algunos pacientes pueden lograr un control de sus síntomas con una dosis de 150 mg administrada cada cuatro semanas. Se recomienda que el médico evalúe periódicamente la necesidad de continuar el tratamiento. Poblaciones especiales:Insuficiencia renal o hepática: No existen estudios del efecto de la insuficiencia renal o hepática en la farmacocinética del omalizumab. Como la depuración del omalizumab, en dosis clínicas, depende principalmente del proceso de depuración de la lgG, por ejemplo, de su degradación en el sistema reticuloendotelial (SRE), es improbable que una insuficiencia renal o hepática pueda alterarla. Mientras no se recomiende un ajuste posológico particular, XOLAIR® deberá ser administrado con precaución en estos pacientes (ver Precauciones generales). Pacientes pediátricos: Cuando un niño alcanza los 12 años de edad la modificación de las dosis queda a juicio del médico: En el asma alérgica, no se ha determinado la inocuidad ni la eficacia del medicamento en pacientes menores de 6 años de edad; por consiguiente, no se recomienda el uso de XOLAIR® en tales pacientes. En la urticaria crónica espontánea, no se ha determinado la inocuidad ni la eficacia del medicamento en pacientes menores de 12 años de edad. Pacientes geriátricos: Existen datos escasos del uso de XOLAIR® en pacientes mayores de 65 años, pero no existe evidencia de que estos pacientes requieran de una dosis diferente que la utilizada en adultos menores de 65 años. Modo de empleo: Instrucciones de uso, manipulación y eliminación del frasco ámpula con polvo liofilizado: XOLAIR® se suministra en viales para uso único y no contiene conservadores antibacterianos. El producto debe utilizarse inmediatamente después de su reconstitución. Se ha demostrado la estabilidad química y física del producto reconstituido durante 8 horas entre 2°C y 8°C y durante 4 horas a 30°C. Desde el punto de vista microbiológico, el producto debe utilizarse inmediatamente después de la reconstitución. Si no se usara de inmediato, el tiempo de conservación durante el uso y las condiciones de conservación antes del uso son responsabilidad del usuario; dicho periodo normalmente no debería ser mayor de 8 horas a temperaturas