ZEMPLAR®
ABBVIE
Cápsulas
Denominación genérica: Paricalcitol.
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: Paricalcitol 1, 2 y 4 mg. Vehículo cbp.
Indicaciones terapéuticas: Paricalcitol está indicado para la prevención y el tratamiento del hiperparatiroidismo secundario asociado con enfermedad renal crónica en etapas 3 y 4 y en pacientes con hiperparatiroidismo secundario asociado con enfermedad renal crónica estadio 5 con hemodiálisis (HD) o diálisis peritoneal (DP).
Farmacocinética y farmacodinamia: Mecanismo de acción: el paricalcitol es un análogo sintético biológicamente activo de la vitamina D del calcitriol con modificaciones a la cadena lateral (D2) y el anillo A (19-nor) lo que permite una activación selectiva del receptor de la vitamina D (RVD). Paricalcitol regula selectivamente el RVD en las glándulas paratiroideas sin incrementar al RVD en el intestino y es menos activo sobre la resorción ósea. Paricalcitol también regula al receptor sensibilizador del calcio (CaSR) en las glándulas paratiroideas. Como resultado de estos efectos, paricalcitol reduce los niveles de hormona paratiroidea al inhibir la proliferación paratiroidea y disminuir la síntesis y secreción de HPT, con un impacto mínimo sobre los niveles de calcio y fósforo, y puede actuar directamente sobre las células óseas para mantener el volumen óseo y mejorar las superficies de mineralización. Al corregir los niveles de HPT anormales, que incluye la normalización de la homeostasis del calcio y fósforo, se puede prevenir o tratar la enfermedad ósea metabólica asociada con la enfermedad renal crónica. Farmacocinética: absorción: paricalcitol se absorbe bien. En sujetos sanos, posterior a la administración oral de paricalcitol a dosis de 0,24 mg/kg, la biodisponibilidad absoluta promedio fue de aproximadamente 72%, la concentración plasmática máxima (Cmáx), el tiempo para lograr la Cmáx (Tmáx) y el área bajo la curva de concentración en el tiempo (AUC0-inf) fueron de 0,630 ng/ml, 3 horas y 5,25 ng•h/ml, respectivamente. La biodisponibilidad absoluta promedio de paricalcitol en pacientes con HD y DP es de 79% y 86%, respectivamente, con una unión elevada, al 95% de intervalo de confianza, de 93% y 112%, respectivamente. Un estudio del efecto de los alimentos en sujetos sanos indicó que la Cmáx y la AUC0-inf se mantuvieron sin cambios cuando paricalcitol se administró con comida con alto contenido de grasas, comparado con el ayuno. Por lo tanto, las cápsulas de paricalcitol pueden ingerirse independientemente de la presencia de alimentos. Distribución: ZEMPLAR® se une extensamente a las proteínas plasmáticas ( > 99%). El volumen aparente de distribución promedio posterior a una dosis de 0,24 mg/kg de paricalcitol en sujetos sanos fue de 34 l. El volumen aparente de distribución promedio posterior a una dosis de 4 mg de paricalcitol en pacientes con ERC Estadio 3 y de 3 mg/kg en pacientes con ERC en Estadio 4 está entre 44 y 46 l. Eliminación: el paricalcitol se elimina primariamente mediante excreción hepatobiliar. En sujetos sanos, la vida media de eliminación promedio es de 5 a 7 horas sobre el rango de dosis estudiada de 0,06 a 0,48 mg/kg. La farmacocinética de la cápsula de paricalcitol ha sido estudiada en pacientes con enfermedad renal crónica (ERC) Estadios 3, 4 y 5. Pacientes en estadio 3 a los que se les administró 4 mg de paricalcitol en cápsulas, la vida media de eliminación es de 17 horas. En pacientes en estadio 4 con ERC, la vida media de eliminación fue de 20 horas cuando se administraron 3 mg de paricalcitol en cápsulas. El grado de acumulación fue consistente con la vida media y la frecuencia de dosificación. El procedimiento de hemodiálisis no tuvo efecto alguno sobre la eliminación de paricalcitol. Metabolismo: posterior a la administración oral de 0,48 mg/kg de una dosis de 3H-paricalcitol, el fármaco inicial fue extensamente metabolizado, alrededor de solo 2% de la dosis fue eliminada sin cambios en heces y no se detectó fármaco inicial en orina. Aproximadamente 70% de la radioactividad se eliminó en heces y 18% fue recuperada en la orina. La mayoría de la exposición sistémica fue a partir del fármaco inicial. Se detectaron dos metabolitos menores, ambos relativos al paricalcitol, en el plasma humano. Un metabolito fue identificado como 24(R)-hidroxi paricalcitol, mientras que el otro metabolito no pudo ser identificado. El 24(R)-hidroxi paricalcitol es menos activo que paricalcitol en un modelo de ratas para la supresión de HTP in vivo. Los datos in vitro sugieren que paricalcitol se metaboliza mediante varias enzimas tanto hepáticas, como extrahepáticas incluyendo el CYP24 mitocondrial, así como CYP3A4 y UGT1A4. Los metabolitos identificados incluyen el producto de hidroxilación 24(R), así como los 24, 26- y 24, 28- de dihidroxilación y de glucuronidación.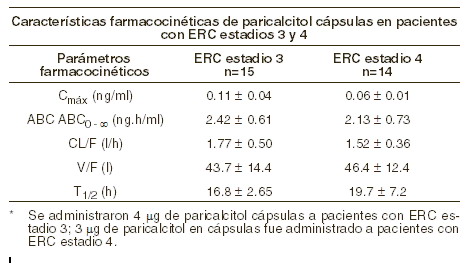
Poblaciones especiales: no se han observado diferencias farmacocinéticas en relación con la edad o el género, en los pacientes adultos estudiados, tampoco se han identificado diferencias raciales. De la misma manera, no se ha estudiado la farmacocinética de paricalcitol en pacientes de menos de 18 años de edad ni en los sujetos de más de 65 años. La farmacocinética de paricalcitol posterior a dosis únicas de rango de dosis de más de 0,06 a 0,48 mcg/kg fueron independientes del género. Alteraciones hepáticas: posterior a la administración intravenosa, la disposición de paricalcitol (0,24 mg/kg) fue comparada en pacientes con alteración hepática leve (n=5) y moderada (n=5), según se indica por el método de Child-Plugh y en sujetos con función hepática normal (n=10). La farmacocinética del paricalcitol no unido fue similar a lo largo del rango de la función hepática evaluada en este estudio. No se requirió de ajuste de la dosis en los pacientes con alteración hepática de leve a moderada. La influencia de la alteración hepática severa sobre la farmacocinética de paricalcitol no ha sido evaluada. Alteraciones renales: posterior a la administración de cápsulas de paricalcitol, el perfil farmacocinético de paricalcitol en sujetos con ERC Estadio 5 con hemodiálisis (HD) o diálisis peritoneal (DP) fue comparable con el de los pacientes con ERC en Estadios 3 y 4. Por lo tanto, no se requiere hacer ajustes especiales de la dosis que aquellas recomendadas en la sección de dosis. Durante la dosificación inicial posterior al ajuste del medicamento, se debe vigilar el calcio, fósforo e HPTi séricos al menos cada dos semanas durante 3 meses después del inicio del tratamiento con paricalcitol o posterior al ajuste de dosis en el tratamiento con paricalcitol, luego monitorear mensualmente durante 3 meses y posteriormente cada 3 meses.
Contraindicaciones: Paricalcitol cápsulas (ZEMPLAR®) no debe administrarse a pacientes con evidencia de toxicidad por vitamina D; hipercalcemia, o hipersensibilidad a cualquiera de los componentes de su fórmula (véase Precauciones generales). Advertencias: el exceso en la supresión de la hormona paratiroidea puede resultar en elevaciones de los niveles de calcio sérico y puede llevar a enfermedad por bajo recambio óseo. Se requiere que a los pacientes se les monitorice la titulación de la dosis para alcanzar los puntos terminales apropiados. La sobredosis aguda por paricalcitol (ZEMPLAR®) puede causar hipercalcemia y requiere atención médica de urgencia. Durante los ajustes de dosis, se deben monitorizar cercanamente los niveles séricos de calcio y fósforo. Si se desarrolla hipercalcemia clínicamente significativa, se debe reducir la dosis o suspender la administración. La administración crónica de paricalcitol (ZEMPLAR®) puede poner a los pacientes en riesgo de hipercalcemia, de elevación del producto Ca x P y de calcificación metastásica. El tratamiento de pacientes con hipercalcemia clínicamente significativa consiste en la reducción inmediata o la interrupción de la administración de paricalcitol (ZEMPLAR®), e incluye una dieta baja en calcio, suspensión de suplementos de calcio, movilización del paciente, tratamiento del desequilibrio hidroelectrolítico, evaluación de anormalidades electrocardiográficas (crítico en pacientes recibiendo digitálicos) y hemodiálisis o diálisis peritoneal, utilizando dializado libre de calcio, como sea necesario. Los niveles séricos de calcio deben monitorizarse frecuentemente, hasta alcanzar normocalcemia. Con paricalcitol (ZEMPLAR®) no deben coadministrarse compuestos que contengan fosfato o vitamina D.
Precauciones generales: La toxicidad por digitálicos se potencia por la hipercalcemia de cualquier causa; de esta forma, se debe tener precaución cuando se prescriban digitálicos junto con paricalcitol (ZEMPLAR®). Si la HPT se suprime alcanzando niveles anormales, pueden desarrollarse lesiones óseas adinámicas (enfermedad por bajo recambio óseo). La vigilancia del paciente y la individualización de la titulación de la dosis es imprescindible para alcanzar los objetivos fisiológicos más apropiados.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: categoría C. Paricalcitol (ZEMPLAR®) mostró causar una discreta disminución de la viabilidad fetal (5%) cuando se administró diariamente a conejas a dosis 0,5 veces la dosis humana de 0,24 mg/kg (en base al área de superficie, mg/m2), y cuando se administró a ratas a dosis 2 veces la dosis humana de 0,24 mg/kg (en base a los niveles plasmáticos de exposición). A las dosis más altas probadas (20 mg/kg, tres veces por semana en ratas, 13 veces la dosis humana de 0,24 mg/kg, en base al área de superficie), hubo un aumento significativo en la mortalidad de las ratas recién nacidas a dosis que fueron tóxicas para la madre (hipercalcemia). No se observaron otros efectos en el desarrollo de los productos de la gestación. A las dosis probadas, el paricalcitol no fue teratogénico. No se han llevado a cabo estudios adecuados y bien controlados en mujeres embarazadas. Paricalcitol (ZEMPLAR®) debe administrarse durante el embarazo únicamente si los beneficios potenciales justifican el riesgo para el feto. Uso durante la lactancia: no se sabe si el paricalcitol se excreta en la leche materna. Debido a que muchos fármacos se excretan en la leche materna, se recomienda precaución cuando se administre paricalcitol (ZEMPLAR®) a una mujer lactando. Uso pediátrico: no se ha establecido la seguridad y la eficacia de las cápsulas de paricalcitol en pacientes pediátricos. La experiencia es limitada con el uso de la inyección de paricalcitol en pacientes menores de 18 años de edad. Uso geriátrico: del número total de pacientes que recibieron paricalcitol en cápsulas (n=220) en los estudios clínicos, 49% fueron de más de 65 años, mientras que 17% tuvieron más de 75 años de edad. Del número total de pacientes con ERC Estadio 5 (n=88) en el estudio pivote de cápsulas de paricalcitol, 28% tuvieron más de 65 años de edad, mientras que 6% tenían más de 75 años. Se observó que no hubo diferencias en cuanto a la seguridad y efectividad entre estos pacientes y en aquellos pacientes jóvenes; de la misma manera, con otras experiencias clínicas reportadas, no se han identificado diferencias en las respuestas entre los pacientes añosos y los jóvenes, pero no se puede descartar una sensibilidad mayor en aquellos individuos con mayor edad.
Reacciones secundarias y adversas: La seguridad de las cápsulas de paricalcitol se ha evaluado en tres estudios controlados de 24 semanas, doblemente a ciegas, controlados con placebo y multicéntricos que involucró a 220 pacientes con ERC estadios 3 y 4. En estos estudios, un total de 107 pacientes recibieron cápsulas de paricalcitol y 113 recibieron placebo. La reacción adversa más comúnmente reportada para los pacientes tratados con paricalcitol fue la erupción cutánea (rash), el cual ocurrió en 2% de los pacientes. La proporción de pacientes que discontinuaron el tratamiento debido a eventos adversos durante los estudios doblemente a ciegas y controlados con placebo fue de 6% para los pacientes tratados con cápsulas de paricalcitol y de 4% para los pacientes tratados con placebo. Esta diferencia no fue estadísticamente significativa. Todos los eventos adversos que fueron al menos posiblemente relacionados con paricalcitol, tanto en ensayos clínicos como en laboratorio se muestran en la siguiente tabla como sistema corporal y su frecuencia. Los siguientes grupos de frecuencia fueron utilizados: muy comunes (≥1/10); comunes (≥1/100 y < 1/10); poco comunes (≥1/1.000 y < 1/100); raros (≥1/10.000 y < 1/1.000); muy raros ( < 1/10.000), incluyendo los reportes aislados.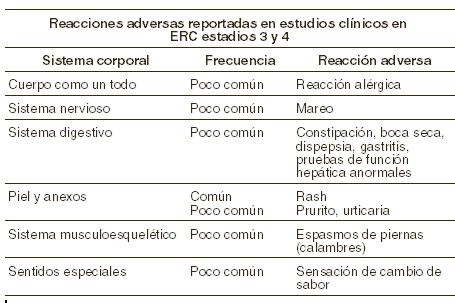
La seguridad de las cápsulas de paricalcitol ha sido evaluada en un estudio clínico multicéntrico, doblemente a ciegas, controlado con placebo de 12 semanas que involucró a 88 pacientes que recibieron cápsulas de paricalcitol y a 27 que recibieron placebo. No hubo diferencias clínicamente importantes ni estadísticamente significativas entre las cápsulas de paricalcitol y placebo en el tipo de incidencias de eventos adversos. La proporción de pacientes que terminaron prematuramente el estudio debido a eventos adversos fue de 7% para los pacientes tratados con cápsulas de paricalcitol y de 7% para los pacientes con placebo. Todos los eventos adversos al menos posiblemente relacionados con las cápsulas de paricalcitol, tanto de manera clínica como en el laboratorio, se muestran en la siguiente tabla por sistema corporal y frecuencia. Los siguientes grupos de frecuencia fueron utilizados: muy comunes (≥1/10); comunes (≥1/100 y < 1/10); poco comunes (≥1/1.000 y < 1/100); raros (≥1/10.000 y < 1/1.000); muy raros ( < 1/10.000), incluyendo los reportes aislados.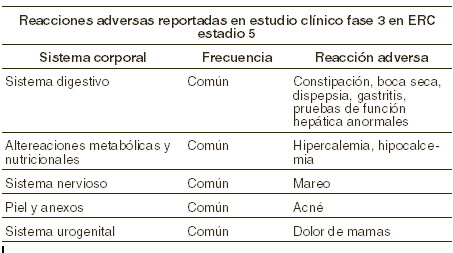
Interacciones medicamentosas y de otro género: Los estudios in vitro indican que paricalcitol no es un inhibidor de CYP3A, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP2E1 en concentraciones de hasta 50 nm (21 ng/ml) (aproximadamente 20 veces mayor que la que se obtiene después de la dosis más alta probada). En hepatocitos primarios frescos cultivados, la inducción observada en las concentraciones de paricalcitol de hasta 50 nm fue menor de dos veces para CYP2B6, CYP2C9 o CYP3A, en los que los controles positivos dieron una inducción de seis a diecinueve veces. Por lo que no se espera que paricalcitol inhiba o induzca la depuración de fármacos metabolizados por estas enzimas. La interacción farmacocinética entre las cápsulas de paricalcitol (16 mg) y omeprazol (40 mg oral) fue investigado en un estudio de dosis única, cruzado en sujetos sanos. La farmacocinética de paricalcitol no se vio afectada cuando se coadministró con omeprazol. El efecto de ketoconazol sobre la farmacocinética de las cápsulas de paricalcitol ha sido estudiada en sujetos sanos. Se afectó mínimamente la Cmáx de paricalcitol, pero el AUC0-a aproximadamente se duplicó en presencia de ketoconazol. La vida media promedio de paricalcitol fue de 17,0 h en presencia de ketoconazol, comparada con 9,8 h, cuando se administró paricalcitol solo.
Alteraciones en los resultados de pruebas de laboratorio: En los estudios controlados con placebo, paricalcitol redujo los niveles séricos de fosfatasa alcalina total.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En un estudio de carcinogenicidad a 104 semanas en ratones CD-1, se observó un aumento en la incidencia de leiomiomas y de leiomiosarcomas uterinos, a dosis subcutáneas (SC) de 1, 3, 10 mg/kg administrados tres veces por semana (2 a 15 veces el ABC a una dosis en humanos de 14 mg, equivalente a 0,24 mg/kg). La incidencia de leiomiomas uterinos fue significativamente diferente del grupo control a la dosis más alta (10 mg/kg). En un estudio de carcinogenicidad a 104 semanas en ratas, hubo un aumento en la incidencia de feocromocitoma suprarrenal benigno a dosis SC de 0,15 a 1,5 mg/kg ( < 1 a 7 veces la exposición posterior a una dosis en humanos de 14 mg, equivalente a 0,24 mg/kg con base a la ABC). El aumento en la incidencia de feocromocitomas en ratas puede relacionarse con la alteración en la homeostasis del calcio debida a paricalcitol. El paricalcitol no exhibió toxicidad genética in vitro con o sin activación metabólica en el estudio de mutagenicidad microbiana (de Ames), en el estudio de mutagenicidad en linfoma de ratón (L5178Y), o en un estudio de aberración cromosómica en células linfocíticas humanas. Tampoco hubo evidencia de toxicidad genética en un estudio in vivo de micronúcleos de ratón, paricalcitol no tuvo efecto sobre la fertilidad (masculina o femenina) en ratas a dosis intravenosas de hasta 20 mg/kg dosis equivalente a 13 veces la dosis máxima recomendada en humanos (0,24 mg/kg en base a la superficie corporal, mg/m2).
Dosis y vía de administración: La dosis se puede administrar independientemente del contenido de alimentos. ERC Estadios 3 y 4: la frecuencia de administración habitual de las cápsulas de paricalcitol es de una cápsula diaria o tres veces por semana (TIW). Cuando se dosifica tres veces por semana, la dosis debería ser administrada con una frecuencia no mayor de cada dos días. Las dosis promedio semanales tanto para el régimen de administración diaria como el de tres veces a la semana son muy parecidas. No se requieren ajustes de dosis en pacientes con daño hepático leve a moderado. Dosis inicial: la dosis inicial de paricalcitol cápsulas está basada en los niveles de hormona paratiroidea intacta (HPTi) basal.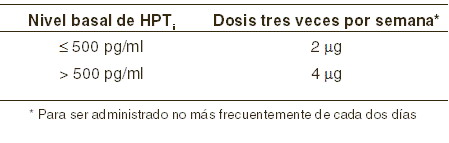
Ajuste de dosis: dosis de mantenimiento: la dosificación debe individualizarse y con base a los niveles de HPTi sérico o plasmático y con una vigilancia sobre el calcio y fósforo sérico. Se sugiere el siguiente esquema de titulación de dosis.
El rango objetivo de niveles de HPT actualmente aceptado en pacientes con insuficiencia renal crónica terminal sometidos a diálisis es no más de 1,5 a 3 veces el límite superior no urémico normal (150-300 pg/ml para HPTi). Se requieren de monitoreo estrecho y del ajuste individual de la dosis, para alcanzar los desenlaces fisiológicos apropiados. Durante cualquier período de ajuste de dosis, se deben monitorizar con mayor frecuencia los niveles séricos tanto de calcio (corregidos para la hipoalbuminemia), como de fósforo. Si se evidencia un nivel de calcio corregido (Ca) ( < 1,2 mg/dl) o un nivel de fósforo (P) elevado ( < 6,5 mg/dl), la dosis de fármaco debe ajustarse, hasta que estos parámetros se normalicen. Si se evidencia hipercalcemia o un producto Ca x P elevado mayor de 65-75 persistente, la dosis del fármaco debe reducirse, o interrumpirse su administración, hasta que estos parámetros se normalicen. Después debe reiniciarse la administración de paricalcitol a una menor dosis. Puede ser necesario disminuir la dosis conforme los niveles de HPT disminuyan en respuesta a la terapia. Así, el ajuste paulatino de la dosis de paricalcitol debe individualizarse. ERC Estadio 5: la frecuencia de administración habitual de las cápsulas de paricalcitol es de una cápsula diaria o tres veces por semana. Cuando se dosifica tres veces por semana, la dosis debería ser administrada con una frecuencia no mayor de cada dos días. Dosis inicial: la dosis inicial de paricalcitol cápsulas está basada en los niveles de hormona paratiroidea intacta (HPTi) basal (en pg/ml)/60. Titulación de la dosis: dosificaciones subsecuentes deberán individualizarse con base a los niveles de HPT, calcio y fósforo séricos. Una titulación de la dosis sugerida de las cápsulas de paricalcitol se basa en la siguiente fórmula: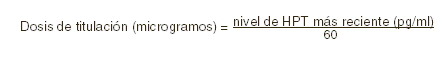
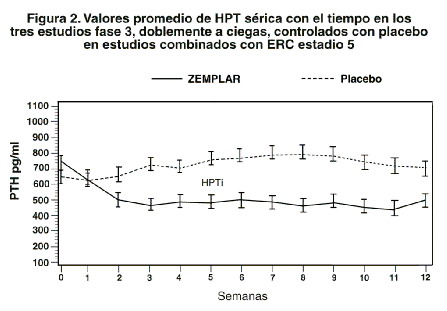
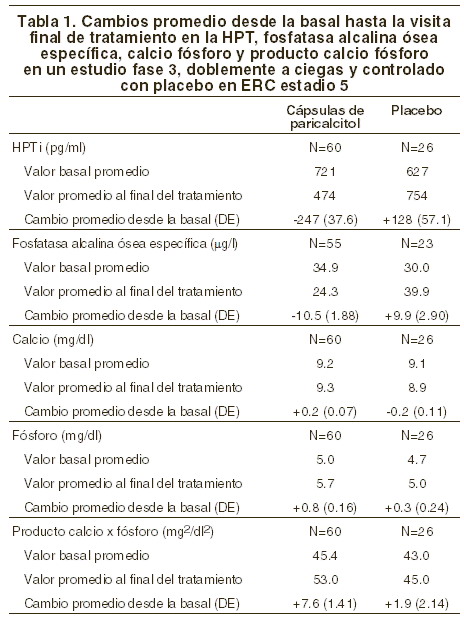
Los niveles de calcio y fósforo séricos deberán vigilarse muy de cerca después del inicio, durante los períodos de titulación de la dosis y durante la coadministración de inhibidores fuertes de P-4503A. Si se observa una elevación del calcio sérico o de Ca x P y el paciente está recibiendo algún quelante de fosfatos a base de calcio, la dosis del quelante debe disminuirse o suspenderse, o el paciente puede cambiarse a un quelante del fósforo que no sea a base de calcio. Si el calcio sérico es > 11,0 mg/dl o la relación Ca x P es > 70 mg2/dl2, la dosis deberá disminuirse en 2 a 4 mg más bajo que el calculado para la TPI/60 más reciente. Si se requiere de un ajuste mayor, la dosis de las cápsulas de paricalcitol deberá reducirse o interrumpirse hasta que estos parámetros se normalicen. Conforme la HPT se acerque al rango normal, pudieran ser necesarias dosis pequeñas e individualizadas con el objeto de lograr una HPT estable. En situaciones en las cuales no se pueda vigilar tan frecuentemente la HPT, el Ca o el fósforo que una vez por semana, se debe iniciar con una dosis inicial más modesta y con una proporción de titulación igualmente modesta. El promedio medio de la dosis TIW durante la semana inicial en el estudio clínico fue de 11,2 mg por dosis. La dosis TIW media promedio general administrada en el estudio clínico fue de 6,3 mg por dosis. La dosis máxima administrada de manera segura en el estudio clínico fue de 32 mg por dosis. Ochenta y dos porciento (82%) de los pacientes tratados con cápsulas de paricalcitol y 78% de los pacientes tratados con placebo terminaron las 12 semanas de tratamiemto. El punto terminal de eficacia primaria de al menos dos reducciones consecutivas de ≥30% de HPTi desde la basal fue lograda por 88% de los pacientes tratados con paricalcitol y en 13% de los pacientes tratados con placebo (p < 0,001). No hubo diferencias estadísticamente significativas entre los pacientes tratados con las cápsulas de paricalcitol y los pacientes tratados con placebo en la incidencia de hipercalcemia (p > 0,999), hiperfosfatemia (p=0,073) o producto de calcio-fósforo elevado (p=0,669). En la basal, 53 (87%) de los sujetos tratados con paricalcitol y 26 (96%) de los sujetos tratados con placebo estaban recibiendo quelantes de fosfatos. En la visita final, la utilización de estos quelantes de fosfatos se mantuvo sin cambios para la mayoría de los sujetos en ambos grupos de tratamiento. Los valores promedio para la HPT, calcio y fósforo séricos con el tiempo, como se muestra en la figura 1, y el patrón de cambio en los valores promedio para la HPTi sérica durante el estudio se muestran en la Figura 2. Los cambios promedio desde la basal hasta la visita final de tratamiento en la HPT, calcio y fósforo séricos y Ca x P se muestran en la tabla de esta misma sección.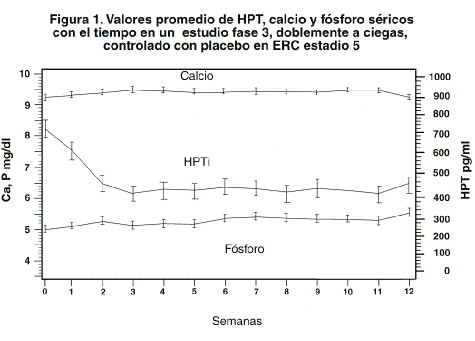
Manifestaciones y manejo de la sobredosificación o ingesta accidental: La sobredosis de paricalcitol (ZEMPLAR®) puede llevar a hipercalcemia (véase Advertencias).
Presentación(es): Frasco con 30 cápsulas de 1, 2 y 4 mg.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 25°C.
Leyendas de protección: No se deje al alcance de los niños. Su venta requiere receta médica. Consérvese en lugar fresco y seco. El empleo de este medicamento durante el embarazo queda bajo la responsabilidad del médico.
Nombre y domicilio del laboratorio: ABBOTT LABORATORIES DE MEXICO, S.A. de C.V., Av. Coyoacán 1622, Col. Del Valle, 03100, México, D.F.
Número de registro del medicamento: 119M2007 SSA IV.
Clave de IPPA: EEAR-06330060102386/R2007.
ZEMPLAR®
ABBVIE
Solución inyectable
Denominación genérica: Paricalcitol.
Forma farmacéutica y formulación: Solución inyectable. Cada ampolleta contiene: Paricalcitol 5 mg. Vehículo cbp 1 ml.
Indicaciones terapéuticas: La inyección de paricalcitol está indicada para la prevención y el tratamiento del hiperparatiroidismo secundario asociado con insuficiencia renal crónica.
Farmacocinética y farmacodinamia: Mecanismo de acción: el paricalcitol es un análogo sintético del calcitriol, forma metabólicamente activa de la vitamina D. La vitamina D y el paricalcitol han demostrado reducir los niveles de hormona paratiroidea. (HPT). Farmacocinética: distribución: el paricalcitol se une extensamente a proteínas plasmáticas (más de 99%). En sujetos sanos, el volumen de distribución en estado estable es de 23,8 l aproximadamente. El volumen promedio aparente de distribución después de una dosis de 0,24 mg/kg en pacientes con insuficiencia renal crónica Estadio 5 que requieren hemodiálisis o diálisis peritoneal es de entre 31 y 35 l. La farmacocinética de paricalcitol ha sido estudiada en pacientes con insuficiencia renal crónica Estadio 5 que requieren hemodiálisis y diálisis peritoneal. La hemodiálisis no tiene efecto en la eliminación de paricalcitol. Sin embargo, comparado con sujetos sanos, los pacientes con insuficiencia renal Estadio 5 muestran una disminución en la aclaración y un incremento en la vida media. Metabolismo: se detectaron varios metabolitos tanto en orina, como en heces, sin haber paricalcitol detectable en orina. Los datos in vitro sugieren que paricalcitol se metaboliza mediante varias enzimas tanto hepáticas como extrahepáticas incluyendo el CYP24 mitocondrial, así como CYP3A4 y UGT1A4. Los metabolitos identificados incluyen el producto de hidroxilación 24(R) (presente en bajos niveles en plasma), así como los 24, 26 y 24, 28 de dihidroxilación y de glucuronidación. El paricalcitol no es inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A a concentraciones de hasta 50 nM (21 ng/ml). Se notó una inducción menor del doble de CYP2B6, CYP2C9 Y CYP3A4 a concentraciones equivalentes de paricalcitol. La unión del paricalcitol a las proteínas plasmáticas in vitro es extensa ( > 99,9%) y no saturable, en el rango de concentración de 1 a 100 ng/ml.
Poblaciones especiales: no se han observado diferencias farmacocinéticas en relación con la edad o el género, en los pacientes adultos estudiados, tampoco se han identificado diferencias raciales. Eliminación: el paricalcitol se elimina primariamente mediante excreción hepatobiliar; 63% de la dosis radio-marcada se recuperó en heces y 19% de la misma se excretó en orina de sujetos sanos. En sujetos sanos la vida media promedio de eliminación es entre 5 a 7 horas después de una dosis de 0,04 a 0,16 mg/kg.
Contraindicaciones: Paricalcitol (ZEMPLAR®) no debe administrarse a pacientes con evidencia de toxicidad por vitamina D, hipercalcemia, o hipersensibilidad a cualquiera de los componentes de su fórmula (véase Precauciones generales). Advertencias: la sobredosis aguda por paricalcitol (ZEMPLAR®) puede causar hipercalcemia y requiere atención médica de urgencia. Durante los ajustes de dosis, se deben monitorizar cercanamente los niveles séricos de calcio y fósforo. Si se desarrolla hipercalcemia clínicamente significativa, se debe reducir la dosis o suspender la administración. La administración crónica de paricalcitol (ZEMPLAR®) puede poner a los pacientes en riesgo de hipercalcemia, de elevación del producto Ca x P y de calcificación metastásica. Los signos y síntomas de intoxicación por vitamina D asociados con hipercalcemia incluyen: a) Tempranos: debilidad, cefalea, somnolencia, náuseas, vómitos, xerostomía, constipación, dolor muscular, dolor óseo y sabor metálico. b) Tardíos: anorexia, pérdida de peso, conjuntivitis (calcárea), pancreatitis, fotofobia, rinorrea, prurito, hipertermia, disminución de la libido, nitrógeno ureico sanguíneo elevado, hipercolesterolemia, AST y ALT elevadas, calcificación ectópica, hipertensión, arritmias cardíacas, somnolencia, muerte y, raramente, psicosis manifiesta. El tratamiento de pacientes con hipercalcemia clínicamente significativa consiste en la reducción inmediata o la interrupción de la administración de paricalcitol (ZEMPLAR®), e incluye una dieta baja en calcio, suspensión de suplementos de calcio, movilización del paciente, tratamiento del desequilibrio hidroelectrolítico, evaluación de anormalidades electrocardiográficas (crítico en pacientes recibiendo digitálicos) y hemodiálisis o diálisis peritoneal, utilizando dializado libre de calcio, como sea necesario. Los niveles séricos de calcio deben monitorizarse frecuentemente, hasta alcanzar normocalcemia. Con paricalcitol (ZEMPLAR®) no deben coadministrarse compuestos que contengan fosfato o vitamina D.
Precauciones generales: La toxicidad por digitálicos se potencia por la hipercalcemia de cualquier causa; de esta forma, se debe tener precaución cuando se prescriban digitálicos junto con paricalcitol. (ZEMPLAR®) Si la HPT se suprime alcanzando niveles anormales, pueden desarrollarse lesiones óseas adinámicas (enfermedad por bajo recambio óseo). El procedimiento de hemodiálisis no tiene efecto en la eliminación de paricalcitol. Sin embargo, comparado con sujetos sanos, los pacientes con insuficiencia renal crónica estadio 5 mostraron una disminución en la aclaración y un incremento en la vida media.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: paricalcitol (ZEMPLAR®) mostró causar una discreta disminución de la viabilidad fetal (5%) cuando se administró diariamente a conejas a dosis 0,5 veces la dosis humana de 0,24 mg/kg, (en base al área de superficie, mg/m2) y cuando se administró a ratas a dosis 2 veces la dosis humana de 0,24 mg/kg (en base a los niveles plasmáticos de exposición) A las dosis más altas probadas, (20 mg/kg, tres veces por semana en ratas, 13 veces la dosis humana de 0,24 mg/kg, en base al área de superficie) hubo un aumento significativo en la mortalidad de las ratas recién nacidas a dosis que fueron tóxicas para la madre (hipercalcemia). No se observaron otros efectos en el desarrollo de los productos de la gestación. A las dosis probadas, el paricalcitol no fue teratogénico. No se han llevado a cabo estudios adecuados y bien controlados en mujeres embarazadas. Paricalcitol (ZEMPLAR®) debe administrarse durante el embarazo únicamente si los beneficios potenciales para la madre, justifican el riesgo para el feto. Uso durante la lactancia: no se sabe si el paricalcitol se excreta en la leche materna. Ya que muchos fármacos se excretan en la leche materna, se recomienda precaución cuando se administre paricalcitol (ZEMPLAR®) a una mujer lactando.
Reacciones secundarias y adversas: Eventos adversos en estudios clínicos fases 2 y 3: en cuatro estudios controlados con placebo, doble ciego, multicéntricos, la descontinuación de la terapia debida a eventos adversos ocurrió en 6,5% de 62 pacientes tratados con paricalcitol y en 2% de 51 pacientes en placebo, durante uno a tres meses. Los eventos adversos que ocurrieron con una frecuencia mayor a 2%, sin importar la causalidad, se presentan en la siguiente tabla:
Si un paciente reportó el mismo término médico más de una vez, se contó una sola vez para el mismo. Los parámetros de seguridad (cambios promedio en Ca, P y Ca x P) durante un estudio de seguridad abierto, de hasta trece meses de duración, apoyan la seguridad a largo plazo del paricalcitol en esta población de pacientes. Eventos adversos en estudios clínicos fase 4: en un estudio fase 4 de hallazgo de dosis, se informó comúnmente de cefalea (2%) y de alteración del gusto (2%). Reacciones adversas de experiencia poscomercialización: durante la experiencia poscomercialización, se ha informado raramente de la aparición de: urticaria, reacción alérgica, edema facial y oral, alteración del gusto (sabor metálico), rash y prurito.
Interacciones medicamentosas y de otro género: Un estudio in vitro indica que el paricalcitol no es un inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A a concentraciones de hasta 50nM (21ng/ml), aproximadamente 20 veces mayor que la obtenida con la dosis más alta probada. En cultivos primarios de hepatocitos frescos, la inducción observada a concentraciones de paricalcitol de hasta 50nM fue menos de 2 veces para CYP2B6, CYP2C9 o CYP3A, en donde los controles positivos tuvieron una inducción de 6 a 19 veces. De aquí que no se espera que paricalcitol inhiba o induzca el aclaramiento de medicamentos metabolizados por estas enzimas. No se han hecho estudios de interacciones medicamentosas con paricalcitol. Aunque no se estudió junto con la inyección de paricalcitol, el efecto de múltiples dosis de ketoconazol administradas 200mg cada 12 horas por 5 días sobre la farmacocinética de las cápsulas de paricalcitol se estudió en sujetos sanos. Se afectó mínimamente la Cmáx de paricalcitol, pero el AUC0-a aproximadamente se duplicó en presencia de ketoconazol. La vida media promedio de paricalcitol fue de 17 h en presencia de ketoconazol, comparada con 9,8 h, cuando se administró solamente el paricalcitol. Dado que el paricalcitol es metabolizado parcialmente por CYP3A y que se sabe que el ketoconazol es un potente inhibidor de la enzima del citocromo P-450 3A, se debe tener precaución al dosificar paricalcitol con ketoconazol y con otros inhibidores de P-450 3A.
Alteraciones en los resultados de pruebas de laboratorio: En los estudios controlados con placebo, paricalcitol redujo los niveles séricos de fosfatasa alcalina total.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En un estudio de carcinogenicidad a 104 semanas en ratones CD-1, se observó un aumento en la incidencia de leiomiomas y de leiomiosarcomas uterinos, a dosis subcutáneas (SC) de 1 a 10 mg/kg, 2 a 15 veces el área bajo la curva a la dosis en humanos de 14 mg equivalente a 0,24 mg/kg. La incidencia de leiomiomas uterinos fue significativamente diferente del grupo control a la dosis más alta (10 mg/kg). En un estudio de carcinogenicidad a 104 semanas en ratas, hubo un aumento en la incidencia de feocromocitoma suprarrenal benigno a dosis SC de 0,15 a 1,5 mg/kg (≤ de 1 a 7 veces la exposición después de una dosis en humanos de 14 mg equivalente a 0,24 mg/kg basado en el área bajo la curva. El aumento en la incidencia de feocromocitomas en ratas puede relacionarse con la alteración en la homeostasis del calcio debida a paricalcitol. El paricalcitol no exhibió toxicidad genética in vitro con o sin activación metabólica en el estudio de mutagenicidad microbiana (de Ames), en el estudio de mutagenicidad en linfoma de ratón (L5178Y), o en un estudio de aberración cromosómica en células linfocíticas humanas. Tampoco hubo evidencia de toxicidad genética en un estudio in vivo de micronúcleos de ratón, paricalcitol no tuvo efecto sobre la fertilidad (masculina o femenina) en ratas a dosis intravenosas de hasta 20 mg/kg dosis (equivalente a 13 veces la dosis en humanos de 14 mg (0,24 mg/kg en base a la superficie corporal, mg/m2).
Dosis y vía de administración: La vía de administración habitual de la inyección de la solución de paricalcitol es a través de la vía utilizada durante la hemodiálisis. Para pacientes sin acceso a hemodiálisis, las inyecciones de paricalcitol deben administrarse mediante inyección intravenosa lenta, durante por lo menos 30 segundos; para minimizar el dolor durante la administración. Adultos: dosis inicial: hay dos métodos alternativos para determinar la dosis inicial de paricalcitol. La dosis máxima administrada de forma segura durante los estudios clínicos fue tan alta como 40 mg. Dosis inicial basada en el peso corporal: la dosis inicial recomendada de paricalcitol es 0,04 mg/kg a 0,1 mg/kg (2,8-7 mg) administrados en bolo IV, con una frecuencia no mayor a cada tercer día, en cualquier momento durante la diálisis. Dosis inicial con base en los niveles basales de HPTi: se ha utilizado el método de HPT intacta (HPTi) como medida de la HPT biológicamente activa en pacientes con insuficiencia renal crónica (IRC). La dosis inicial se calcula mediante de la siguiente fórmula, administrándose como bolo intravenoso, no más frecuentemente de cada tercer día, en cualquier momento durante la diálisis:
Ajuste de dosis: el rango objetivo de niveles de HPT actualmente aceptado en pacientes con insuficiencia renal crónica terminal sometidos a diálisis es no más de 1,5 a 3 veces el límite superior no urémico normal (150-300 pg/ml para HPTi). Se requieren de monitoreo estrecho y del ajuste individual de la dosis, para alcanzar los desenlaces fisiológicos apropiados. Durante cualquier período de ajuste de dosis, se deben monitorizar con mayor frecuencia los niveles séricos tanto de calcio (corregidos para la hipoalbuminemia), como de fósforo. Si se evidencia un nivel de calcio corregido mayor a 11,2 mg/dl o un nivel de fósforo elevado mayor a 6,5 mg/dl), la dosis del fármaco debe ajustarse, hasta que estos parámetros se normalicen. Si se evidencia hipercalcemia o un producto Ca x P elevado mayor de 75 persistente, la dosis del fármaco debe reducirse, o interrumpirse su administración, hasta que estos parámetros se normalicen. Después debe reiniciarse la administración de paricalcitol a una menor dosis. Puede ser necesario disminuir la dosis conforme los niveles de HPT disminuyan en respuesta a la terapia. Así, el ajuste paulatino de la dosis de paricalcitol debe individualizarse. Si no se observa una respuesta satisfactoria, la dosis puede aumentarse de 2 a 4 mg, a intervalos de dos a cuatro semanas. Si en cualquier mom
