ZIAGENAVIR®
GSK
Solución oral
Denominación genérica: Abacavir.
Forma farmacéutica y formulación: Solución oral. Solución. Cada 100 mL contienen: Sulfato de abacavir equivalente a 2 g de abacavir. Vehículo cbp 100 mL.
Indicaciones terapéuticas: ZIAGENAVIR® está indicado en la terapia antirretroviral combinada de la infección por el virus de la inmunodeficiencia humana (VIH) en niños y adultos.
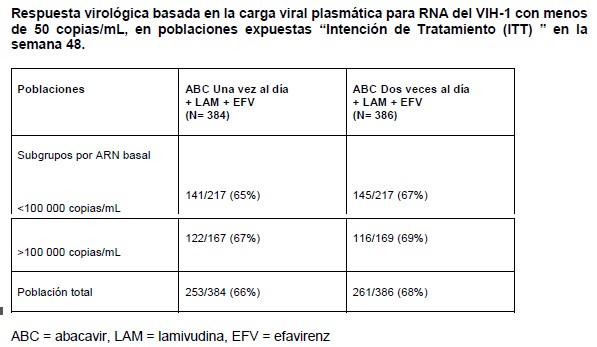
Farmacocinética y farmacodinamia: Farmacocinética: Absorción: ZIAGENAVIR® se absorbe rápida y adecuadamente después de su administración oral con una biodisponibilidad absoluta en adultos de alrededor del 83%. El tiempo promedio (tmáx) para alcanzar la máxima concentración sérica del abacavir es de 1.5 hrs. para la tableta y 1.0 hrs. para la solución, después de su administración oral. No se han observado diferencias en el área bajo la curva (ABC) entre la tableta y la solución. A una dosis de 300 mg 2 veces al día, la Cmáx media en estado de equilibrio para las tabletas de abacavir fue de aproximadamente 3.00 mg/mL y el promedio del ABC con un intervalo de dosificación de cada 12 hrs. es de aproximadamente 6.02 mg.h/mL (ABC diaria de aproximadamente 12.0 mcg/.h/ml). El valor de la Cmáx para la solución oral es ligeramente mayor que el de la tableta. Después de una tableta de 600 mg, la Cmáx media de abacavir fue de aproximadamente 4.26 mcg//ml y el promedio del ABC fue de 11.95 mcg.h/ml. La administración con alimentos retrasó la absorción y disminuyó la Cmáx pero no afectó la concentración plasmática total (ABC), por lo que ZIAGENAVIR® puede ser administrado con ó sin alimentos. Distribución: Después de la administración intravenosa, el volumen de distribución aparente fue de alrededor de 0.8 L/Kg, lo que indica que el abacavir penetra libremente en los tejidos corporales. Los estudios en pacientes infectados por el VIH han demostrado un buen paso del abacavir al líquido cefalorraquídeo (LCR), con una proporción en el ABC de LCR/plasma 30 a 44%. En un estudio de farmacocinética fase I, la penetración del abacavir al LCR fue investigada después de la administración de 300 mg dos veces al día. La concentración media de abacavir alcanzada en el LCR después de 1.5 hrs. fue de 0.14 mg/mL. En un estudio farmacocinético ulterior con 600 mg dos veces al día, la concentración de abacavir en LCR se incrementó con el tiempo, desde aproximadamente 0.13 mg/mL en 0.5 a 1 h, después de administrar la dosis, hasta aproximadamente 0.74 mg/mL después de 3 a 4 hrs. Aunque las concentraciones máximas pudieron no haberse alcanzado a las 4 hrs., los valores observados son 9 veces mayores que la CI50 de abacavir de 0.08 mg/mL ó 0.26 mM. Los estudios in vitro, indican que el abacavir se fija en grado bajo a moderado (49%) nivel a las proteínas plasmáticas humanas a concentraciones terapéuticas. Esto indica una baja probabilidad de interacciones con otras drogas por desplazamiento de la fijación a proteínas plasmáticas. Metabolismo: El abacavir es metabolizado principalmente por el hígado y cerca del 2% de la dosis administrada es excretada sin cambios por vía renal. Las principales vías metabólicas en el humano ocurren por la deshidrogenasa alcohólica y la glucuronidación para producir el ácido 5'-carboxílico y 5'-glucurónido respectivamente, los cuales cuentan cerca del 66% de la dosis administrada. Estos metabolitos son excretados en la orina. Eliminación: La vida media promedio del abacavir es de aproximadamente 1.5 hrs. Después de administrar varias dosis orales de 300 mg de abacavir 2 veces al día no hay evidencia significativa de acumulación del compuesto. La eliminación es por vía del metabolismo hepático con la excreción subsecuente de los metabolitos principalmente por la orina. Dichos metabolitos y una cantidad de abacavir inalterado constituyen cerca del 83% de la dosis administrada de abacavir que aparece en orina, el resto se elimina a través de las heces fecales. Grupos Especiales de Pacientes: Niños: El abacavir se absorbe óptima y rápidamente a partir de formulaciones de una solución oral y tableta administrada a niños. Se ha demostrado que la exposición de abacavir en plasma es la misma para ambas formulaciones cuando se administran a la misma dosis. A los niños a los que se les administra abacavir en solución oral conforme al régimen de dosificación recomendado alcanzan una exposición a abacavir en plasma semejante a la de los adultos. A los niños a los que se les administra abacavir en tabletas orales conforme al régimen de dosificación recomendado alcanzan una exposición abacavir en plasma mayor que la de los niños a los que se les administra solución oral debido a que se administran dosis mayores en mg/kg con la formulación en tableta (véase Dosis y Administración). Estudios de farmacocinética pediátrica demostraron que una dosis al día provee el equivalente a la dosis de dos veces al día ABC0-24 de la misma dosis total diaria para ambas formulaciones solución oral y tabletas. No hay suficientes datos de seguridad para recomendar el uso de ZIAGENAVIR® en lactantes menores de 3 meses de edad. Los datos limitados de los que se dispone indican que una dosis de 2 mg/Kg administrada a neonatos menores de 30 días de edad proporciona niveles ABC similares, o mayores, a los de la dosis de 8 mg/Kg administrada a niños de mayor edad. Pacientes de edad avanzada: La farmacocinética del abacavir no ha sido estudiada en pacientes mayores de 65 años de edad. En el tratamiento de éstos pacientes, debe considerarse la mayor frecuencia de disminución de las funciones hepática, renal y/o cardiaca, enfermedades concomitantes u otra terapia medicamentosa. Insuficiencia Renal: El abacavir es metabolizado principalmente a través del hígado y cerca del 2% es excretado sin cambios en la orina. La farmacocinética del abacavir en pacientes con enfermedad renal terminal es similar a la que se observa en los pacientes con función renal normal. Por lo tanto no se requiere reducción en la dosis en los pacientes con daño renal. Insuficiencia Hepática: El abacavir es metabolizado principalmente a través del hígado. La farmacocinética del abacavir ha sido estudiada en pacientes con insuficiencia hepática leve (Child-Pugh de 5 a 6). Los resultados muestran un incremento promedio de 1.89 veces en el ABC y de 1.58 veces la vida media de abacavir. El ABC de los metabolitos no fue modificado por la hepatopatía, sin embargo fueron reducidas las tasas de formación y de eliminación de éstos. Con la finalidad de lograr exposiciones en el rango terapéutico de los pacientes sin daño hepático, los pacientes con hepatopatía leve deben recibir 200 mg de abacavir dos veces al día. No se ha estudiado la farmacocinética del abacavir en casos de hepatopatías de moderada a severa, por lo que el abacavir no se recomienda en estos grupos de pacientes. Estudios clínicos: En un estudio doble ciego de más de 48 semanas de tratamiento en pacientes adultos sin tratamiento previo, el uso de abacavir, zidovudina y lamivudina mostró un efecto equivalente al del uso combinado de indinavir, lamivudina y zidovudina en el análisis primario de eficacia. En un análisis secundario, los pacientes con más de 100 000 copias/mL de RNA del VIH-1 que recibieron la combinación con indinavir mostraron una respuesta superior. Los pacientes con niveles basales menores a 100 000 copias/mL, mostraron una respuesta equivalente a ambos tratamientos. El régimen de abacavir + lamivudina administrados una vez al día se investigó en un estudio multicéntrico, controlado, doble ciego (CNA 30021) con 770 pacientes adultos sin tratamiento previo e infectados por VIH-1. Los pacientes fueron distribuidos aleatoriamente para recibir ya fuera ZIAGENAVIR® a dosis de 600 mg administrados una vez al día, ó 300 mg administrados dos veces al día, ambos en combinación con 300 mg de lamivudina, administrados una vez al día, y 600 mg de efivarenz, administrados una vez al día. Los pacientes se estratificaron en función del nivel basal de ARN del VIH-1 del plasma menor o igual a 100, 000 copias/mL o mayores de esta cifra. La duración del tratamiento doblemente ciego fue de 48 semanas por lo menos y los resultados aparecen en la siguiente tabla.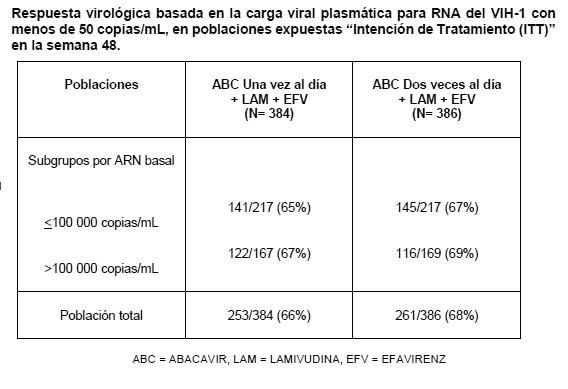
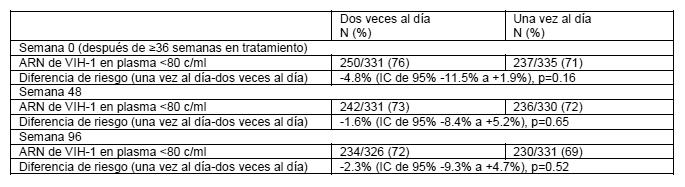
El grupo tratado con abacavir una vez al día no fue inferior cuando se contrastó con el tratado en total con este medicamento dos veces al día y la carga viral basal de los subgrupos respectivos. La incidencia de eventos adversos fue similar en ambos grupos de tratamiento. Se intentó realizar un análisis genotípico de los pacientes con falla virológica (confirmada por carga viral > 50 copias/mL). Se registró una menor falla virológica en los pacientes con ambos tratamientos de una y dos veces al día (10 y 8% respectivamente). Adicionalmente el genotipo se restringió a muestras de plasma con más de 500 copias de ARN del-VIH-1/ mL. Esta condición resultó en una muestra pequeña, cuyos resultados no confirmaron las diferencias existentes en las mutaciones emergentes de estos dos grupos de tratamiento. Los residuos de 184 de la transcriptasa reversa fueron consistentes con las mutaciones asociadas a los ITRAN (M184V o 184I). La segunda mutación más frecuente fue la L74V. Las mutaciones Y115F y K65R fueron raras. Se estableció una comparación de asignación aleatoria de un régimen que incluía dosificación una versus dos veces al día de abacavir y lamivudina dentro de un estudio controlado de asignación aleatoria y multicéntrico de pacientes pediátricos infectados con VIH. Se reclutaron 1,206 pacientes pediátricos con edades entre 3 meses y 17 años en el Ensayo ARROW (COL105677) y se les dosificó de conformidad con las recomendaciones de rango de peso en los lineamientos de la Organización Mundial de la Salud (Terapia antirretroviral de la infección por VIH en infantes y niños, 2006). Después de 36 semanas en un régimen que incluyó abacavir y lamivudina dos veces al día, 669 sujetos elegibles fueron asignados de manera aleatoria a una dosificación continua dos veces al día o se les cambió a abacavir y lamivudina por al menos 96 semanas. Los resultados se resumen en la tabla a continuación: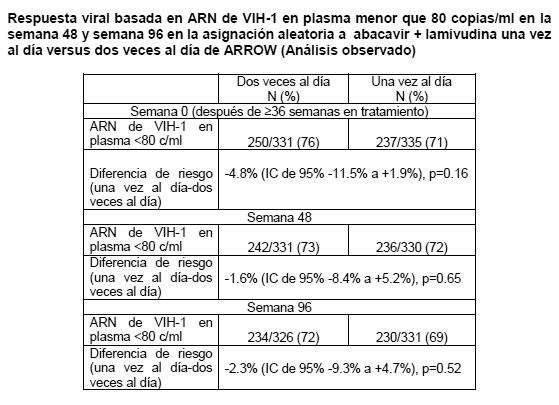
Se demostró que el grupo de dosificación de abacavir/lamivudina una vez al día no era inferior al grupo de dos veces al día de conformidad con el margen de no inferioridad previamente especificado de -12%, para el criterio de valoración principal de < 80 c/ml en la Semana 48 así como en la Semana 96 (criterio de valoración secundario) y todos los demás límites evaluados ( < 200c/ml, < 400c/ml, < 1000c/ml), los cuales se ubicaron claramente dentro de este margen de no inferioridad. Las pruebas de análisis de subgrupo en cuanto a heterogeneidad de una versus dos veces al día no demostraron efecto significativo del sexo, la edad o la carga viral en la asignación aleatoria. Las conclusiones respaldaron la no inferioridad indistintamente del método de análisis. En un estudio abierto para los inhibidores nucleósidos de la transcriptasa reversa (INTR), (con o sin cegamiento para nelfinavir) en niños, una mayor proporción de los tratados con abacavir y lamivudina (73%) ó abacavir y zidovudina (70%) tuvieron igual ó menos de 400 copias/mL de RNA VIH-1 a las 24 semanas, comparados con aquellos tratados con lamivudina y zidovudina (44%). En niños con importante exposición a antivirales, fue observado un modesto pero sostenido efecto de la combinación abacavir, lamivudina y zidovudina. En el tratamiento de pacientes experimentados, el grado beneficio a partir de la adición de abacavir dependerá de la naturaleza y duración de la terapia previa que pudiera haberse seleccionado para tratar la resistencia cruzada al abacavir. Registro Antirretroviral en el Embarazo: El Registro Antirretroviral en el Embarazo ha recibido reportes prospectivos de más de 2,000 exposiciones a abacavir durante el embarazo que terminaron en nacimientos vivos. Estos consistieron en más de 800 exposiciones durante el primer trimestre, mas de 1,100 exposiciones durante el segundo / tercer trimestres e incluyeron 27 y 32 defectos al nacimiento respectivamente. La prevalencia (95% IC) de defectos en el primer trimestre fue 3.1% (2.0, 4.4%) y en el segundo/tercer trimestres, 2.7% (1.9, 3.9%). Entre las embarazadas en la población de referencia, la incidencia considerada habitual de defectos al nacimiento es de 2.7%. No hubo asociación entre abacavir y defectos al nacimiento en general observados en el Registro del uso de antiretrovirales en el embarazo. Farmacodinamia: Grupo farmacoterapeútico: análogo de nucleósido, código ATC: J05A F06. Mecanismo de acción: El abacavir es un inhibidor de la transcriptasa reversa, análogo nucleósido (ITRAN). Es un potente antiviral contra los VIH-1 y VIH-2, incluyendo las cepas de VIH-1 resistentes a zidovudina, lamivudina, zalcitabina, didanosina y nevirapina. El abacavir es metabolizado intracelularmente a su molécula activa, carbovir 5'-trifosfato (TP). Los estudios in vitro han mostrado que inhibe a la enzima transcriptasa reversa del virus del VIH, lo que resulta en la terminación de la cadena y la interrupción del ciclo de replicación viral. La actividad antiviral de abacavir en cultivos celulares no fue antagonizada cuando se combina con los inhibidores nucleósidos de la transcriptasa reversa (NRTIs) didanosina, emtricitabina, lamivudina, stavudina, tenofovir, zalcitabina o zidovudina, los inhibidores no nucleósidos de la transcriptasa reversa (NNRTI) nevirapina o el inhibidor de la proteasa (PI) amprenavir. En un estudio de 20 pacientes infectados que recibieron ZIAGENAVIR® 300 mg dos veces al día, con una dosis tomada antes del muestreo del período de 24 hrs., el promedio de la vida media terminal del carbovir trifosfato en el estado de equilibrio fue de 20.6 hrs. comparado con el promedio geométrico de la vida media del abacavir en plasma en este estudio de 2.6 hrs. En estado de equilibrio las propiedades farmacocinéticas de ZIAGENAVIR® 600 mg una vez al día se compararon con ZIAGENAVIR® 300 mg dos veces al día en un estudio cruzado con 27 pacientes infectados por VIH. Las exposiciones a trifosfato de carbovir en sangre periférica de las células mononucleares fueron más elevadas con ZIAGENAVIR® 600 mg una vez al día el ABC24,ss(32 %, más elevada), Cmax 24,ss (99% más elevada) y los valores inferiores (18% más elevados), en comparación con la dosis de 300 mg dos veces al dia. Adicionalmente la eficacia y la seguridad de ZIAGENAVIR® administrado una vez al día se demostró en un estudio pivote (CNA 30021- ver estudios clínicos). Se han seleccionado in vitro cepas del VIH-1 resistentes al abacavir, asociadas con cambios genotípicos específicos en la transcriptasa reversa (codones M184V, K65R, L74V y Y115F). La resistencia viral al abacavir se desarrolla en forma relativamente lenta in vitro e in vivo, requiriendo múltiples mutaciones para alcanzar un incremento de 8 veces más en la CI50 con respecto al virus primario (o silvestre) original, lo que puede ser clínicamente importante. Las cepas resistentes al abacavir pueden también mostrar una sensibilidad disminuida a la lamivudina, zalcitabina y/o didanosina, pero permanecen sensibles a la zidovudina y la estavudina. Existe poca probabilidad de resistencia cruzada entre el abacavir y los IP's o inhibidores no nucleósidos de la transcriptasa reversa (INNTR). La falla al tratamiento posterior a la terapia inicial con abacavir, lamivudina y zidovudina principalmente, se asocia solo con la mutación M184V, manteniendo así opciones terapéuticas para una segunda línea de tratamiento. El abacavir penetra al líquido cefalorraquídeo (LCFR) y se ha demostrado que reduce la carga viral en él. En combinación con otros agentes ARV's puede tener un papel en la prevención de las complicaciones neurológicas del VIH y puede retrasar la aparición de resistencias en este sitio de refugio.
Contraindicaciones: ZIAGENAVIR® está contraindicado en pacientes con hipersensibilidad conocida al abacavir o a cualquier otro componente de las tabletas o la solución.
Precauciones generales: Reaccion de hipersensibilidad. (Ver Reacciones Secundarias y Adversas). Abacavir está asociado con un riesgo de reacciones de hipersensibilidad (RHS) caracterizadas con fiebre y/o erupción cutánea con otros síntomas que indican una implicación de múltiples órganos. Las RHS pueden poner en riesgo la vida y en casos inusuales pueden ser mortales cuando no se tratan correctamente. El riesgo de que ocurra una RHS por abacavir aumenta significativamente en pacientes con resultados positivos en la prueba del alelo HLA-B*5701. No obstante, las RHS por abacavir se han reportado con menor frecuencia en pacientes que no poseen este alelo. Se debería cumplir con lo siguiente: La evaluación del estado de HLA-B*5701 debería considerarse antes de iniciar el tratamiento con abacavir y también antes de reiniciar el tratamiento con abacavir en pacientes con estado HLA-B*5701 desconocido que toleraron el abacavir previamente. ZIAGENAVIR® no se recomienda para su uso en pacientes con el alelo HLA- B*5701 ni es pacientes con sospecha previa de HSR por abacavir mientras tomaban cualquier otro medicamento con abacavir (e.g. KIVEXA, TRIZIVIR, TRIUMEQ) sin importar la condición de HLA-B*5701. Se le deberá recordar a cada paciente leer el instructivo para el paciente incluido en el empaque de ZIAGENAVIR®. Se les debería recordar la importancia de tomar la Tarjeta de alerta incluida en el empaque y conservarla en todo momento. En cualquier paciente tratado con ZIAGENAVIR®, el diagnóstico clínico de sospecha de reacción de hipersensibilidad debe ser la base de la toma de decisiones clínicas. ZIAGENAVIR® debe detenerse sin retraso, incluso en la ausencia del alelo HLA- B*5701, si se sospecha de RHS. El retraso para detener el tratamiento con ZIAGENAVIR® después del inicio de la hipersensibilidad podría resultar en una reacción de riesgo para la vida. Se les deberá indicar a los pacientes que han experimentado reacciones de hipersensibilidad, eliminar sus tabletas restantes de ZIAGENAVIR® con el fin de evitar reiniciar el abacavir. Reiniciar productos que contienen abacavir después de RHS por abacavir puede resultar en un reinicio rápido de los síntomas en horas y podría incluir hipotensión de riesgo para la vida y muerte. Sin importar el estado de HLA-B*5701 del paciente, si se ha descontinuado el tratamiento con cualquier producto que contiene abacavir por cualquier razón y reiniciar el tratamiento con abacavir está en consideración, la razón para descontinuar debe establecerse. Si RHS no puede descartarse, ZIAGENAVIR® o cualquier otro medicamento que contenga abacavir (e.g. KIVEXA®) no debe reiniciarse. Si se descarta una reacción de hipersensibilidad, los pacientes pueden reiniciar el ZIAGENAVIR®. Pocas veces, los pacientes que detuvieron el tratamiento con abacavir por otras razones además de los síntomas de RHS también experimentaron reacciones que ponen en peligro su vida a horas de reiniciar el tratamiento con abacavir (ver la Sección 4.8 Descripción de reacciones adversas seleccionadas). Los pacientes deben estar conscientes de que RHS puede ocurrir al reiniciar ZIAGENAVIR® o cualquier otro medicamento que contenga abacavir (e.g. KIVEXA®) y que reiniciar el ZIAGENAVIR® o cualquier otro medicamento que contenga abacavir (e.g. KIVEXA®) debería asumirse sólo si puede accederse rápidamente a la atención médica. Descripción clínica de RHS por abacavir: Las RHS por abacavir se han caracterizado bien por medio de estudios clínicos y durante el seguimiento posterior a la comercialización. Los síntomas suelen aparacer durante las primeras seis semanas (tiempo medio de inicio 11 días) del inicio del tratamiento con abacavir, aunque estas reacciones pueden ocurrir en cualquier momento durante el tratamiento. Casi todas las RHS por abacavir incluyen fiebre o exantema, o ambas cosas, como parte del síndrome. Otros signos y síntomas que se han observado como parte de las RHS por abacavir incluyen síntomas respiratorios y gastrointestinales, que podrían llevar a un diagnóstico equivocado de RHS como enfermedad respiratoria (neumonía, bronquitis, faringitis) o gastroenteritis (ver Reacciones adversas, Descripción de reacciones adversas seleccionadas). Los síntomas relacionados con RHS empeoran si se continúa con la terapia y pueden llegar a ser potencialmente mortales. Estos síntomas suelen resolverse al suspender la terapia con ZIAGENAVIR®. Acidosis Láctica/ Hepatomegalia severa con esteatosis: Se ha informado de casos de acidosis láctica y la hepatomegalia severa con esteatosis, incluyendo casos fatales, con el empleo de antirretrovirales análogos nucleósidos ya sea solos o en combinación, incluyendo abacavir, en el tratamiento de la infección por VIH. La mayoría de estos casos han ocurrido en mujeres. Los datos clínicos característicos del desarrollo de acidosis láctica, son debilidad generalizada, anorexia y pérdida de peso súbita e inexplicable, así como síntomas gastrointestinales y respiratorios (disnea y taquipnea). Se debe tener precaución cuando se administre abacavir a cualquier paciente y particularmente a aquellos con riesgo conocido de enfermedad hepática. El tratamiento con abacavir se debe suspender en pacientes que hayan desarrollado datos clínicos ó de laboratorio de acidosis láctica o hepatotoxicidad (hepatomegalia y esteatosis hepática aún en ausencia de elevación de las aminotransaminasas). Lípidos séricos y glucosa en sangre: Los niveles de lípidos séricos y glucosa en sangre pueden aumentar durante la terapia antirretroviral. El control de la enfermedad y los cambios en el estilo de vida también pueden ser factores contribuyentes. Se debe tener en consideración la medición de los niveles de lípidos séricos y glucosa en sangre. Las alteraciones de lípidos deben ser manejadas apropiadamente de acuerdo a la clínica. Síndrome de Reconstitución Inmunológica: En los pacientes infectados por VIH con severa deficiencia inmune al inicio de su tratamiento antirretroviral (ARV), puede ocurrir una reacción inflamatoria por infecciones oportunistas residuales y causar severo deterioro clínico o agravamiento de los síntomas. Típicamente estas reacciones se han observado en las primeras semanas o meses del inicio del tratamiento ARV. Ejemplos importantes de estos casos son: retinitis por citomegalovirus, infecciones micobacterianas generalizadas o focales (o ambas) y neumonías focales o diseminadas por Pneumocystis jiroveci (A menudo referido como PCP.) Cualquier síntoma inflamatorio debe evaluarse sin demora e iniciarse el tratamiento cuando sea necesario. Se ha reportado la presentación de enfermedades autoimmunes (tales como la enfermedad de Graves, polimiositis y el síndrome de Guillain-Barre) durante las fases iniciales de la reconstitución inmunitaria, sin embargo, el tiempo de aparición es mas variable, y puede ocurrir muchos meses después del inicio del tratamiento y algunas veces pueden ser de presentación atípica. Infecciones oportunistas: Los pacientes que reciben ZIAGENAVIR® o cualquier otra terapia antirretroviral pueden desarrollar infecciones oportunistas y otras complicaciones de la infección por VIH. Por lo tanto deben permanecer bajo estrecha observación clínica por un médico experto en el tratamiento de enfermedades asociadas al VIH. Transmisión de la infección: Se debe advertir a los pacientes que no se ha probado que la terapia ARV actual, incluyendo ZIAGENAVIR®, evite el riesgo de transmisión del VIH a otros sujetos a través del contacto sexual o contaminación de la sangre. Se deben seguir tomando precauciones adecuadas Infarto del Miocardio: Varios estudios epidemiológicos de carácter observacionales, han informado de una asociación con el uso de abacavir y el riesgo de infarto de miocardio. Los metaanálisis de ensayos controlados aleatorios no han observado ningún exceso de riesgo de infarto de miocardio con el uso de abacavir. Hasta ahora no existe un mecanismo biológico establecido para explicar un incremento potencial del riesgo. Todos los datos disponibles a partir de estudios observacionales y estudios clínicos controlados muestran inconsistencia y por lo tanto la evidencia de una relación causal entre el tratamiento con abacavir y el riesgo de infarto de miocardio no es concluyente. Solución oral: La formulación ZIAGENAVIR® en solución oral contiene sorbitol, el cual es capaz de ocasionar dolor abdominal y diarrea. Al metabolizarse, el sorbitol se convierte en fructosa, por lo que no es adecuado administrar esta formulación a pacientes con intolerancia hereditaria a la fructosa. Efectos sobre la capacidad de conducir y operar máquinas: No hay datos disponibles en la actualidad que sugieran que ZIAGENAVIR® afecta la capacidad de conducir vehículos u operar maquinaria.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Abacavir se ha evaluado en el Registro del uso de antirretrovirales en el embarazo en más de 2000 mujeres durante el embarazo y postparto. La información en humanos disponible en el Registro de Antirretrovirales en el Embarazo no demuestra un aumento del riesgo de defectos mayores al nacimiento por abacavir comparado con la incidencia considerada habitual (véase Estudios Clínicos). Sin embargo, no hay estudios adecuados y bien controlados en embarazadas y no se ha establecido el uso seguro de ZIAGENAVIR® en el embarazo humano. El abacavir se ha relacionado con algunos hallazgos en estudios reproductivos en animales, sin embargo estos estudios no son siempre predictivos de la respuesta en humanos, por lo que la administración de ZIAGENAVIR® durante el embarazo debe considerarse sólo cuándo el beneficio para la madre supere el riesgo potencial para el producto. Lactancia: Estudios en ratas han mostrado que el abacavir y sus metabolitos son secretados en la leche materna en el período de lactancia. Es previsible que ocurra lo mismo en el género humano, aunque esto no ha sido confirmado. No existen datos disponibles sobre la seguridad de ZIAGENAVIR® administrado en lactantes menores de 3 meses de edad. Por otro lado algunos expertos recomiendan que las madres infectadas por el VIH no alimenten al seno materno a sus infantes bajo ninguna circunstancia con objeto de evitar la transmisión del VIH; lo que refuerza la recomendación de que las madres lactantes no alimenten al seno a sus infantes mientras reciban tratamiento con ZIAGENAVIR®. Se han reportado elevaciones transitorias y leves del lactato sérico, las cuales pueden deberse a disfunción mitocondrial en neonatos y lactantes expuestos in utero o periparto a ITRAN. Se desconoce la relevancia clínica de esta anomalía. En muy raras ocasiones también se han reportado retrasos en el desarrollo, convulsiones y otras alteraciones neurológicas. No se ha establecido relación causal alguna entre estos eventos y la exposición a ITRAN, in utero o periparto. Estos hallazgos no afectan las recomendaciones actuales del uso de ARV's en las pacientes embarazadas, para prevenir la transmisión vertical del VIH. Expertos en salud recomiendan que, cuando se posible, las madres infectadas con el VIH no amamanten a sus lactantes, con el fin de evitar la transmisión del VIH. En las ocasiones en que la alimentación con fórmula no sea posible, deben seguirse las guías locales oficiales de lactancia y tratamiento al considerar el amamantamiento durante la terapia antirretroviral. En un estudio después de la administración oral repetida de 300 mg de abacavir dos veces al día (dados como TRIZIVR), la relación leche materna: plasma materno fue 0.9. La mayoría de los lactantes (8 de 9) tuvieron niveles de abacavir no detectables (sensibilidad de la prueba 16 ng/mL). Los niveles intracelulares de trifosfato de carbovir (metabolito activo de abacavir) en infantes en lactancia no se midieron por lo que se desconoce la relevancia clínica de las concentraciones séricas de los compuestos originales.
Reacciones secundarias y adversas: En el caso de muchos de los otros eventos adversos reportados, es incierta su relación con ZIAGENAVIR® ya sea por los otros medicamentos empleados en el tratamiento del SIDA o como resultado de la enfermedad misma. Muchos de los eventos adversos listados a continuación (náusea, vómito, diarrea, fiebre fatiga y exantema) se presentan comúnmente como parte de los síntomas de la RHS al abacavir. En consecuencia los pacientes con alguno de estos síntomas, deben ser cuidadosamente evaluados para descartar o establecer la presencia de una RHS. Si se había suspendido el ZIAGENAVIR® y posteriormente se toma la decisión de continuarlo, esto sólo debe hacerse bajo estricta vigilancia médica. (ver Precauciones generales - Consideraciones especiales después de interrumpir el tratamiento). La mayoría de los eventos adversos que se mencionan a continuación, no han limitado el uso del ZIAGENAVIR®. Se ha utilizado la siguiente escala para clasificarlos: Muy comunes: > 1/10. Comunes: > 1/100 y < 1/10. No comunes: > 1/1000 y < 1/100. Raros: > 1/10 000 y < 1/1000. Muy raros: < 1/10 000. Datos de Estudios Clínicos: Alteraciones del metabolismo y nutricionales: Común: Anorexia. Alteraciones del sistema nervioso: Común: Cefalea. Alteraciones gastrointestinales: Comunes: Náusea, vómito y diarrea. Alteraciones generales y/o relacionadas con la vía de administración: Comunes: Fiebre, letargo, fatiga. En los estudios clínicos controlados, las alteraciones de laboratorio relacionadas con ZIAGENAVIR® fueron raras, sin diferencias en la incidencia observada entre los pacientes tratados con ZIAGENAVIR® y los controles. Población pediátrica: La base de datos de seguridad para respaldar la dosificación una vez al día de abacavir en pacientes pediátricos proviene del Ensayo ARROW (COL105677) en el que se le administró abacavir y lamivudina una o dos veces al día a 669 sujetos pediátricos infectados con VIH-1 (véase Estudios clínicos). No se han identificado problemas adicionales de seguridad en los sujetos pediátricos a los que se les administró la dosificación una o dos veces al día en comparación con los adultos. Estudios post-comercialización (PMS) o de Farmacovigilancia: Alteraciones de Metabolismo y de la nutrición: Comunes: Hiperlactatemia. Raros: Acidosis láctica (ver Precauciones Generales). Alteraciones gastrointestinales: Raros: Se ha reportado pancreatitis, pero la relación causal al tratamiento con ZIAGENAVIR® es incierta. Alteraciones en piel y tejido subcutáneo: Comunes: Exantema (sin síntomas sistémicos). Muy raros: Eritema multiforme, síndrome de Stevens Johnson y necrolisis epidérmica tóxica. Descripción de reacciones adversas seleccionadas. Hipersensibilidad (ver también Advertencias y precauciones). La reacción de hipersensibilidad (RHS) por abacavir se ha identificado como una reacción adversa común con el tratamiento con abacavir. Los signos y síntomas de esta reacción de hipersensibilidad se describen a continuación. Se han identificado ya sea por estudios clínicos como por vigilancia posterior a la comercialización. Los reportados en al menos 10% de los pacientes con una reacción de hipersensibilidad se encuentran en negritas. Casi todos los pacientes que desarrollan reacciones de hipersensibilidad tendrán fiebre y/o erupción (usualmente maculopapular o de urticaria) como parte del síndrome, sin embargo, las reacciones han ocurrido sin erupción ni fiebre. Otros síntomas clave incluyen síntomas gastrointestinales, respiratorios o de constitución como letargo y malestar. Piel: Erupción (usualmente maculopapular o urticaria). Tracto gastrointestinal: Náuseas, vómito, diarrea, dolor abdominal, úlceras bucales. Vías respiratorias: Disnea, tos, garganta irritada, síndrome de dificultad respiratoria del adulto, falla respiratoria. Misceláneos: Fiebre, fatiga, malestar, edema, linfadenopatía, hipotensión, conjuntivitis, anafilaxia. Neurológico/ psiquiátrico: Dolor de cabeza, parestesias. Hematológico: Linfopenia. Hígado/páncreas: Elevación de las pruebas de funcionamiento hepático, insuficiencia hepática. Musculo esquelético: Mialgia, raramente miólisis, artralgia, elevación de la creatina fosfoquinasa (CPK). Urología: Creatinina elevada, insuficiencia renal. Reiniciar el abacavir después de una RHS por abacavir resulta en un regreso temprano de los síntomas en horas. Esta reaparición del RHS suele ser más grave que en la presentación inicial y podría incluir hipotensión que pone en peligro la vida y muerte. Además, las acciones no han ocurrido con frecuencia después de reiniciar el abacavir en pacientes que sólo tuvieron uno de los síntomas claves de la hipersensibilidad (ver previamente) antes de detener al abacavir y en ocasiones inusuales también se ha notado en pacientes que reiniciaron el tratamiento sin síntomas anteriores de RHS (i.e., pacientes ya considerados como tolerantes al abacavir). Para obtener los detalles de la gestión clínica en el caso de una RHS sospechosa por abacavir ver Advertencias y precauciones.
Interacciones medicamentosas y de otro género: Con base en estudios in vitro, el potencial para interacciones medicamentosas es bajo. El abacavir no tiene el potencial para inhibir el metabolismo mediado por la vía de la enzima 3A4 del citocromo oxidasa P450 y tampoco se ha observado que interactúe in vitro con medicamentos metabolizados por las enzimas CYP 3A4, CYP2C9 ó CYP2D6. No se ha observado inducción del metabolismo hepático. En consecuencia existe poco potencial para interacciones con IP's y otras drogas metabolizadas por el sistema enzimático P450. Los estudios clínicos muestran que no existen interacciones significativas entre el abacavir, la zidovudina y la lamivudina. Efecto de Abacavir sobre la Farmacocinética de Otros Agentes: Abacavir in vitro, demostró una débil o ninguna inhibición del transportador de aniones orgánicos 1B1 (OATP1B1), OATP1B3, la proteína resistente de cáncer de mama (BCRP) o P-glicoproteína (Pgp) y una mínima inhibición del transportador de catión orgánico 1 (OCT1), OCT2 y la proteína de toxina de extrusión a múltiples fármacos 2-K (MATE2-K). Por lo tanto no se espera que el Abacavir afecte las concentraciones plasmáticas de fármacos que son sustratos de estos transportadores. Abacavir es un inhibidor in vitro de MATE1; Sin embargo, tiene un bajo potencial para afectar las concentraciones plasmáticas de los sustratos MATE1 en exposiciones terapéuticas de medicamentos (de hasta 600 mg). Efecto de Otros Agentes sobre la Farmacocinética de Abacavir: Abacavir in vitro no es un sustrato de OATP1B1, OATP1B3, OCT1, OCT2, OAT1, Mate1, MATE2-K, a la proteína de múltiples fármacos asociada a la resistencia 2 (MRP2) o MRP4, por lo tanto, no se espera que los fármacos que modulan estos transportadores afecten las concentraciones plasmáticas del abacavir. A pesar de abacavir es un sustrato de BCRP y Pgp in vitro, los estudios clínicos demuestran ningún cambio clínicamente significativas en la farmacocinética de abacavir cuando se administra conjuntamente con lopinavir / ritonovir (inhibidores de Pgp y BCRP). Interacciones relevantes de abacavir: Etanol: El metabolismo del abacavir se altera por la administración concomitante de etanol, lo cual resulta en aumento en el ABC del abacavir en cerca del 41%. Dado el perfil de seguridad del abacavir, dichos hallazgos se consideran clínicamente insignificantes. El abacavir no tiene efecto sobre el metabolismo del etanol. Metadona: En un estudio farmacocinético, la coadministración de 600 mg de abacavir dos veces al día con metadona, demostró una reducción del la Cmáx del abacavir en un 35% y una hora de retraso en el tmáx, pero el ABC se mantuvo sin cambios. Los cambios en la farmacocinética del abacavir no se consideran clínicamente significativos. En este estudio el abacavir incrementó el aclaramiento promedio de la metadona a un 22%. Este cambio no se considera clínicamente significativo para la mayoría de los pacientes, sin embargo ocasionalmente se podría necesitar un ajuste de la dosis de la metadona. Retinoides: Los compuestos retinoides tales como la isotretinoina, se eliminan por la vía de la deshidrogenasa alcohólica. Es posible la interacción con abacavir, pero aún no se ha estudiado.
Alteraciones en los resultados de pruebas de laboratorio: En estudios clínicos controlados, las anormalidades de laboratorio relacionadas con ZIAGENAVIR® no fueron comunes. No hubo diferencia en cuanto a incidencia entre los pacientes tratados con ZIAGENAVIR® y los del grupo control.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad, mutagenicidad: El abacavir no fue mutagénico en pruebas bacterianas, pero mostró actividad in vitro en el análisis de aberración cromosómica del linfocito humano, el análisis del linfoma del ratón y la prueba del micronúcleo in vivo. Esto coincide con la actividad conocida de otros análogos de nucleósido. Estos resultados indican que el abacavir es un clastógeno débil, tanto in vitro como in vivo, a concentraciones altas para pruebas. Los estudios sobre carcinogenicidad con abacavir administrado oralmente en ratones y ratas, mostraron un incremento en la incidencia de tumores malignos y no malignos. Los tumores malignos se produjeron en las glándulas prepuciales de los machos y las glándulas clitorianas de las hembras, en ambas especies, y en el hígado, vejiga urinaria, ganglios linfáticos y la subdermis en ratas hembras. La mayoría de estos tumores se presentó a la dosis más alta de abacavir, consistente en 330 mg/Kg/día, en ratones, y en 600 mg/Kg/día en ratas. Estos niveles de dosificación fueron equivalentes a 24 - 32 veces la exposición sistémica humana esperada. La excepción fue el tumor de las glándulas prepuciales, que se produjo a una dosis de 110 mg/Kg. Esto es equivalente a seis veces la exposición sistémica esperada en el ser humano. No existe una contraparte estructural de estas glándulas en la especie humana. Aunque se desconoce el potencial carcinogénico en los seres humanos, estos datos sugieren que el riesgo carcinogénico en los humanos es excedido por el beneficio clínico potencial. Teratogénesis: Se ha visto que se produce una transferencia placentaria de abacavir, de sus metabolitos relacionados, o de ambos, en los animales. Solo se encontraron indicios de toxicidad, en el desarrollo del embrión y el feto, en ratas que recibieron dosis maternas tóxicas de 500 mg/Kg/día, y mayores. Esta dosis equivale a 32 - 35 veces la exposición terapéutica humana basada en el ABC. Los hallazgos incluyeron edema fetal, variaciones y malformaciones, resorciones, disminución del peso corporal fetal, así como un aumento en el número de partos de productos muertos. La dosis con la cual no se presentaron efectos en el desarrollo pre o posnatal, consistió en 160 mg/Kg/día. Esta dosis