ONGLYZA
ABBOTT
Denominación genérica: Saxagliptina.
Forma farmacéutica y formulación: Tabletas. Formulación: Cada tableta contiene: Clorhidrato de saxagliptina equivalente a 2.5 mg y 5.0 mg de saxagliptina. Excipiente cbp Una tableta.
Indicaciones terapéuticas: Monoterapia: ONGLYZA® está indicada como terapia complementaria a la dieta y el ejercicio para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2. Terapia en Combinación: Combinación adicionada: ONGLYZA® está indicada en pacientes con diabetes mellitus tipo 2 para mejorar el control glucémico en combinación con metformina, una tiazolidinediona (TZD) una sulfonilurea (SU) o con insulina (con o sin metformina), cuando con el uso de estos agentes como monoterapia, junto con la dieta o ejercicio, no se ha logrado un control glucémico adecuado. Combinación inicial: ONGLYZA® está indicada como terapia inicial en combinación con metformina, como complemento a la dieta y al ejercicio, para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2 cuando la terapia doble con saxagliptina y metformina es apropiada. ONGLYZA® no debe ser utilizada en pacientes con diabetes tipo 1 o para el tratamiento de cetoacidosis diabética.
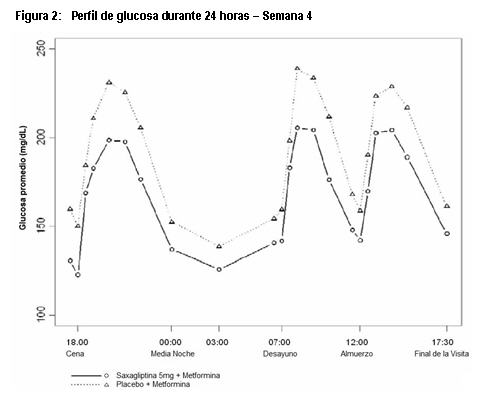
Farmacocinética y farmacodinamia: Mecanismo de Acción: Saxagliptina es un inhibidor de la enzima Dipeptidilpeptidasa 4 (DPP-4); altamente competitivo, selectivo, reversible y potente. Saxagliptina demuestra selectividad para DPP4 versus otras enzimas DPP, incluyendo DPP8 y DPP9. Saxagliptina tiene una unión prolongada al sitio activo de DPP4, ampliando su inhibición. Saxagliptina ejerce su acción en pacientes con diabetes tipo 2 al retardar la inactivación de las hormonas incretinas, incluyendo el péptido 1 similar al glucagon (GLP-1) y el polipéptido insulinotrópico dependiente de glucosa (GIP). Las concentraciones de estas hormonas incretinas activas intactas, son incrementadas por saxagliptina, con lo cual se aumenta y prolonga su acción. Las hormonas incretinas son liberadas por el intestino durante el día y las concentraciones se incrementan en respuesta a los alimentos. Estas hormonas son rápidamente inactivadas por la enzima DPP4. Las incretinas son parte de un sistema endógeno que está involucrado en la regulación fisiológica de la homeostasis de la glucosa. Cuando se elevan las concentraciones de glucosa en sangre, GLP-1 y GIP incrementan la síntesis y la liberación de insulina a partir de las células beta pancreáticas. GLP-1 también disminuye la secreción de glucagon a partir de las células alfa pancreáticas, permitiendo reducir la producción de glucosa hepática. Las concentraciones de GLP-1 están reducidas en pacientes con diabetes tipo 2, pero saxagliptina incrementa GLP-1 y GIP activos, potenciando estos mecanismos. Al incrementar y prolongar las concentraciones de incretina activa, saxagliptina incrementa la liberación de insulina y disminuye las concentraciones de glucagon en la circulación, de una manera dependiente de la glucosa. ONGLYZA® mejora el control glucémico al reducir las concentraciones de glucosa en ayuno y postprandiales en pacientes con diabetes tipo 2, al mejorar la función de las células alfa y beta, como se refleja en las acciones descritas más adelante. Secreción de insulina dependiente de la glucosa en ayuno. ONGLYZA® incrementa la capacidad de respuesta de las células beta pancreáticas hacia la glucosa en ayuno y conduce a un aumento en la secreción de insulina y a la disposición de glucosa en presencia de concentraciones elevadas de glucosa. Secreción de insulina postprandial, dependiente de glucosa: ONGLYZA® incrementa la capacidad de respuesta de las células beta pancreáticas a la glucosa en el estado postprandial y conduce a una secreción aumentada de la insulina postprandial y al aumento en la disposición de glucosa. Secreción de glucagon postprandial: En diabetes tipo 2, los incrementos paradójicos en la secreción de glucagon a partir de las células alfa después de tomar los alimentos, estimula la producción de glucosa hepática y contribuye a la disregulación glucémica. ONGLYZA® modera la secreción de glucagon y disminuye las concentraciones de glucagon postprandial. Farmacodinamia: Mejora en el control glucémico: En pacientes con diabetes tipo 2, la administración de ONGLYZA® condujo a la inhibición de la actividad enzimática de DPP4 por un periodo de 24 horas. Después de una carga de glucosa oral o una comida, esta inhibición de DPP4 dio como resultado un incremento de 2 a 3 veces en los niveles circulantes de GLP-1 y GIP activos, disminución de la concentración de glucagon e incremento en la capacidad de respuesta de las células beta dependientes de glucosa, que resultó en concentraciones más altas de insulina y de péptido C. La elevación de la insulina y la disminución en el glucagon estuvieron asociadas con una reducción en el nivel de glucosa en ayuno y con una reducción en la excursión de glucosa posterior a una carga oral de glucosa o la ingesta de un alimento. El tratamiento con saxagliptina 5 mg y metformina de liberación prolongada una vez al día con los alimentos vespertinos por 4 semanas, produjo una reducción significativa en la concentración promedio de glucosa durante las 24 horas del intervalo de dosificación (definido como el área bajo la curva de glucosa durante 24 horas dividida entre 24 horas) en comparación con placebo más metformina de liberación prolongada (reducción media placebo corregida de -16.8 mg/dL, p=0.0001), con mejoras consistentes en los valores de medición de glucosa en plasma durante las 24 horas del intervalo de dosificación (Figuras 1 y 2). Se observaron reducciones significativas en la glucosa postprandial a las dos horas y en la glucosa plasmática en ayuno de dos días (reducción media placebo corregida de -35.4 mg/dL, p=0.0010 y -15.3 mg/dL, p=0.0002, respectivamente).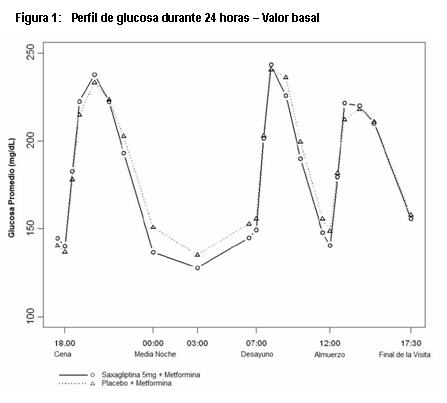
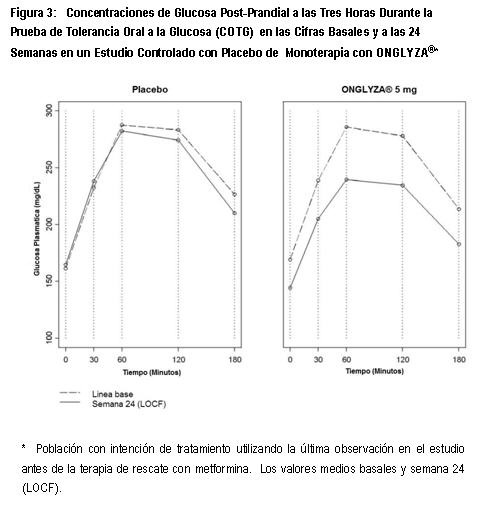
Electrofisiología Cardiaca: En un estudio clínico diseñado para estudiar el efecto de ONGLYZA® en el intervalo QTc, la dosificación con ONGLYZA® no estuvo asociada con la prolongación clínicamente significativa del intervalo QTc o del ritmo cardiaco a dosis diarias hasta de 40 mg (8 veces la Dosis Humana Recomenda (RHD)). En un estudio comparativo activo, cruzado de cuatro vías, controlado con placebo, doble ciego, aleatorizado, a 40 sujetos sanos les fueron administradas dosis de hasta 40 mg de saxagliptina o placebo una vez al día por cuatro días, o dosis única de moxifloxacina de 400 mg como control positivo. Después de la dosis de 40 mg, el incremento máximo en los cambios medios ajustados por placebo en el intervalo QTc y el ritmo cardiaco desde los basales, fueron 2.4 mseg a las 24 horas después de la dosis y 4.5 latidos por minuto a las 4 horas después de la dosis, respectivamente. Farmacocinética: La farmacocinética de saxagliptina ha sido ampliamente caracterizada en sujetos sanos y en pacientes con diabetes tipo 2. Saxagliptina fue rápidamente absorbida después de la administración oral, alcanzando usualmente concentraciones plasmáticas máximas (Cmax) dentro de las dos horas posteriores a la administración en ayuno. Los valores de Cmax y del área bajo la curva (AUC) aumentaron de manera proporcional al incremento en la dosis de saxagliptina. Después de una dosis oral de 5 mg de saxagliptina a sujetos sanos, los valores plasmáticos medios de AUC INF para saxagliptina y su principal metabolito activo fueron de 78 ng·h/mL y 214 ng·h/mL, respectivamente. Los valores de Cmax en plasma correspondientes fueron de 24 ng/mL y de 47 ng/mL, respectivamente. Los coeficientes de variación intra-sujetos para la Cmax y AUC de saxagliptina fueron menores al 12%. Después de una dosis oral única de 5 mg de saxagliptina a sujetos sanos, la vida media terminal en plasma (t1/2) para saxagliptina fue de 2.5 horas y el valor medio de t1/2 para la inhibición de DPP4 en plasma fue de 27 horas. La inhibición de la actividad de DPP4 en plasma por saxagliptina por al menos 24 horas después de la administración oral de ONGLYZA® es debida a la alta potencia, alta afinidad y unión prolongada al sitio activo. No se observó ninguna acumulación apreciable con la dosificación repetida de una vez al día a cualquier nivel de dosis. No se observó ninguna dependencia a la dosis y al tiempo en la depuración de saxagliptina y su metabolito principal después de 14 días de dosificación una vez al día con saxagliptina a dosis que van desde 2.5 mg hasta 400 mg. Los resultados del modelo de exposición basado en la población, sugieren que la farmacocinética de saxagliptina y su metabolito activo fueron similares en sujetos sanos y en pacientes con diabetes tipo 2. Absorción: ONGLYZA®puede ser administrada con y sin alimentos. La cantidad de saxagliptina absorbida después de una dosis oral es de al menos 75%. Los alimentos tuvieron efectos relativamente modestos sobre la farmacocinética de saxagliptina en los sujetos sanos. La administración con una comida alta en grasas dio como resultado ausencia de cambio en la Cmax de saxagliptina y un incremento del 27% en AUC en comparación con el estado en ayuno. El tiempo para que saxagliptina alcance la Cmax (Tmax) fue incrementado por aproximadamente 0.5 horas con alimento en comparación con el estado en ayuno. Estos cambios no fueron considerados clínicamente significativos. Distribución: El enlace de la proteína in vitro de saxagliptina y su metabolito principal en suero humano está por debajo de niveles cuantificables. De este modo, los cambios en los niveles de proteína sanguínea en diversos estados de enfermedad (por ejemplo, insuficiencia renal o hepática) no se espera que alteren la disposición de saxagliptina. Metabolismo: El metabolismo de saxagliptina es mediado principalmente por el citocromo P450 3A4/5 (CYP3A4/5). El metabolito principal de saxagliptina es también un inhibidor de DPP-4 competitivo, reversible, selectivo, la mitad de potente que saxagliptina. Excreción: Saxagliptina es eliminada por vía renal y hepática. Después de una dosis única de 50 mg de 14C-saxagliptina, 24%, 36% y 75% de la dosis fueron excretadas en la orina como saxagliptina, su metabolito principal de la radioactividad total, respectivamente. La depuración renal promedio de saxagliptina (aproximadamente 230 mL/minuto) fue mayor que la tasa de filtración glomerular estimada (aproximadamente 120 mL/minuto), sugiriendo cierta excreción renal activa. Para el metabolito principal, los valores de depuración renal fueron comparables a la tasa de filtración glomerular estimada. Se recuperó un total de 22% de la radioactividad administrada en las heces, lo que representa la fracción de la dosis de saxagliptina excretada en bilis y/o el fármaco no absorbido desde el tracto gastrointestinal. Farmacocinética del Metabolito principal: Los valores de Cmax y AUC para el metabolito principal de saxagliptina se incrementaron de manera proporcional al incremento en la dosis de saxagliptina. Después de dosis orales de 2.5 mg a 400 mg de saxagliptina después de recibir alimento o en ayuno, los valores de AUC medios para el metabolito principal estuvieron en el intervalo de 2 y 7 veces más alto que las exposiciones a saxagliptina original en una base molar. Después de una dosis oral única de 5 mg de saxagliptina en el estado de ayuno, el valor medio de la vida media terminal (t1/2) para el metabolito principal fue de 3.1 horas, y no se observó acumulación apreciable después de la dosificación repetida de una vez al día a cualquier dosis. Poblaciones especiales: Insuficiencia renal: Se realizó un estudio abierto, de dosis única, para evaluar la farmacocinética de saxagliptina (dosis de 10 mg) en sujetos con diversos grados de insuficiencia renal crónica, en comparación a los sujetos con función renal normal. El grado de deterioro renal no afectó la Cmax de saxagliptina o su metabolito principal. En sujetos con CrCL > 50mL/min (correspondiente a TFG estimada ≥ 45mL/min/1.73m2 por la ecuación de MDRD TFG estimada), los valores de AUC de saxagliptina y su metabolito principal fueron 1.2 y 1.7 veces mayores, respectivamente, que los valores de AUC en sujetos con función renal normal. Debido a que los incrementos de esta magnitud no son clínicamente relevantes, el ajuste de la dosis en estos pacientes no es recomendado. En sujetos con insuficiencia renal con CrCL 50mL/min (correspondiente a < 45mL/min/1.73m2) o en sujetos con ESRD en hemodiálisis, los valores de AUC de saxagliptina y su metabolito principal fueron hasta 2.1 y 4.5 veces más altos, respectivamente, que los valores de AUC en sujetos con función renal normal. En estos pacientes, la dosis recomendada es de 2.5 mg una vez al día (Ver Dosis y Vía de Administración y Precauciones generales). Insuficiencia Hepática: No existieron diferencias clínicamente significativas en la farmacocinética para sujetos con insuficiencia hepática leve, moderada o severa, por tanto, no se recomienda ningún ajuste de la dosis para ONGLYZA®, en pacientes con insuficiencia hepática. En sujetos con insuficiencia hepática (clases A, B y C de Child-Pugh) la Cmax media y el AUC de saxagliptina fueron hasta 8% y 77% mayores, respectivamente, en comparación a los controles sanos equivalentes, posterior a la administración de una dosis única de 10 mg de saxagliptina. Los valores de Cmax y AUC correspondientes al metabolito principal fueron hasta 59% y 33% menores, respectivamente, en comparación a los controles sanos pareados. Estas diferencias no son consideradas como clínicamente significativas. Índice de Masa Corporal: No se recomienda ningún ajuste de la dosis con base en el índice de masa corporal (IMC). El IMC no fue identificado como una covariable significativa sobre la depuración aparente de saxagliptina o su metabolito principal, en un análisis de modelo de exposición. Geriátrico: No se recomienda ningún ajuste de la dosis de saxagliptina con base en la edad únicamente. Los sujetos de edad avanzada (entre 65 a 80 años) tuvieron valores de media geométrica más altos de Cmax y valores de media geométrica de AUC de 23% y 59% respectivamente para saxagliptina original en comparación a los sujetos jóvenes (18 a 40 años). Las diferencias en la farmacocinética del metabolito principal entre sujetos de edad avanzada y sujetos jóvenes, reflejaron en general las diferencias observadas en la farmacocinética de saxagliptina original. La diferencia entre la farmacocinética de saxagliptina y el metabolito principal en sujetos jóvenes y sujetos de edad avanzada es probable que se deba a múltiples factores que incluyen la declinación de la función renal y de la capacidad metabólica con el incremento de la edad. La edad no fue identificada como una covariable significativa sobre la depuración aparente de saxagliptina y su metabolito principal, en un análisis de modelo de exposición. Pediátrico y adolescente: La farmacocinética no ha sido estudiada en la población pediátrica, ni en adolescentes menores de 18 años, por lo que no se recomienda. Género: No se recomienda ningún ajuste de la dosis con base en el género. No existieron diferencias observadas en la farmacocinética de saxagliptina entre hombres y mujeres. En comparación a los hombres, las mujeres tuvieron valores de exposición aproximadamente 25% más altos para el metabolito principal que los hombres, pero es improbable que esta diferencia sea de relevancia clínica. El género no fue identificado como una covariable significativa sobre la depuración aparente de saxagliptina y su metabolito principal en un análisis modelo de exposición. Raza: No se recomienda ningún ajuste de la dosis con base en la raza. Un análisis de modelo de exposición comparó la farmacocinética de saxagliptina y su metabolito principal en 309 sujetos blancos con 105 sujetos no blancos (consistentes de 6 grupos raciales). No se detectó ninguna diferencia significativa en la farmacocinética de saxagliptina y su metabolito principal entre estas dos poblaciones. Información de estudios clínicos: Mejora en el control glucémico: ONGLYZA® ha sido estudiada como monoterapia y en combinación con metformina, glibenclamida, tiazolidinedionas, pioglitazona y rosiglitazona e insulina. Se estudiaron 4148 pacientes con diabetes tipo 2 distribuidos aleatoriamente, incluyendo 3021 pacientes tratados con ONGLYZA®, en seis estudios de seguridad y eficacia clínica, controlados, doble ciego, realizados para evaluar los efectos de ONGLYZA® sobre el control glucémico. En estos estudios, la edad media de los pacientes fue de 54 años y 71% de los pacientes eran blancos, 16% eran asiáticos, 4% eran negros, 9% eran de otros grupos raciales. Cuatrocientos veintitrés (423) pacientes adicionales, incluyendo 315 que recibieron ONGLYZA®, participaron en un estudio de intervalo de dosis, controlado con placebo, de 6 a 12 semanas de duración. En estos seis estudios doble ciego, ONGLYZA® fue evaluada la dosis de 2.5 mg, 5 mg y 10 mg una vez al día. El tratamiento con ONGLYZA® en todas las dosis produjo disminuciones clínicamente relevantes y estadísticamente significativas en la hemoglobina glucosilada (HbA1c), la glucosa plasmática en ayuno (GPA), y la glucosa postprandial (GPP), incluyendo la GPP a las 2 horas después de la prueba de tolerancia oral a la glucosa (COTG), en comparación al control. Se observaron reducciones en HbA1c a través de los subgrupos incluyendo género, edad, raza e índice de masa corporal (IMC) basal. En general, la dosis diaria de 10 mg de saxagliptina no proporcionó mayor eficacia que la dosis diaria de 5 mg. Una dosis diaria de 5 mg de ONGLYZA® proporcionó en general mayor reducción en HbA1c y GPP en comparación a la dosis diaria con 2.5 mg de ONGLYZA®. En cuatro estudios adicionales, ONGLYZA® fue evaluada en pacientes con diabetes tipo 2: un estudio controlado con activo versus glipizida, en 858 pacientes inadecuadamente controlados con metformina en monoterapia, un estudio con insulina controlado con placebo en 455 pacientes inadecuadamente controlados únicamente con insulina o con insulina en combinación con metformina un estudio placebo-controlado en 257 pacientes con control glucémico inadecuado con metformina más sulfonilurea y en un estudio controlado con placebo en 170 pacientes con un inadecuado control glucémico e insuficiencia renal (moderada, severa y en etapa terminal en hemodiálisis [ESRD]). Mejora en el control glucémico - monoterapia: Un total de 766 pacientes nunca antes tratados, con diabetes tipo 2 controlada inadecuadamente, participaron en dos estudios controlados con placebo, doble ciego, de 24 semanas de duración para evaluar la eficacia y seguridad de ONGLYZA® en monoterapia. Un total de 401 pacientes nunca antes tratados con diabetes controlada inadecuadamente (HbA1c ≥7% hasta ≤10%) participaron en un estudio controlado con placebo, doble ciego, con una duración de 24 semanas para evaluar la eficacia y la seguridad de la monoterapia con ONGLYZA®. Después de un periodo de 2 semanas con cegamiento, dieta, ejercicio y placebo, los pacientes fueron distribuidos aleatoriamente a recibir dosis de 2.5 mg, 5 mg ó 10 mg de ONGLYZA® o de placebo. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante el estudio fueron tratados con la terapia de rescate con metformina, adicionada a placebo o a ONGLYZA®. La titulación de la dosis de ONGLYZA®no fue permitida en este estudio. El tratamiento con la dosis diaria de 5 mg de ONGLYZA® proporcionó mejoras significativa en HbA1c, GPA y GPP en comparación al placebo (Tabla 1). Se observaron reducciones en HbA1c en la Semana 4 y GPA en la Semana 2 en el grupo de tratamiento con 5 mg de ONGLYZA®, con relación al placebo, en las etapas más tempranas de la evaluación. La proporción de pacientes que lograron HbA1c < 7% (no obstante el valor basal) fue significativamente mayor en el grupo con 5 mg de ONGLYZA® en comparación con el grupo placebo. Se observó una mejoría significativa en el AUC de GPP en 3 horas después de la prueba de toleracia oral a la glucosa (COTG) en el grupo de tratamiento con 5 mg de ONGLYZA®, en comparación con el placebo (Figura 3). Se observaron reducciones significativas en el nivel de GPP a las 2 horas en el grupo de tratamiento con 5 mg de ONGLYZA® (-43 mg/dL) en comparación con -6 mg/dL en el grupo de placebo. La proporción de los pacientes que discontinuaron por falta de control glucémico o que fueron rescatados para cumplir los criterios glucémicos pre-especificados fue más alta en el grupo de placebo (26%) que en el grupo de tratamiento con 5 mg de ONGLYZA® (20%). Un valor basal más alto de HbA1c estuvo asociado con un mayor cambio ajustado a la media, a partir del valor basal en HbA1c, con ONGLYZA® 5 mg. El efecto de ONGLYZA® sobre los criterios de valoración de los lípidos en este estudio fue similar al placebo. ONGLYZA® no incrementó el peso corporal desde la basal (-0.05 kg), en comparación con la pequeña reducción desde el peso basal en pacientes tratados con placebo (-1.35 kg). Estudios de extensión controlado a largo plazo: Los pacientes que fueron rescatados (basado en los niveles de glucosa predefinidos) durante las 24 semanas del periodo inicial del estudio, así como aquéllos que completaron todas las visitas durante las 24 semanas del periodo inicial del estudio sin necesidad de terapia de rescate, fueron elegibles para entrar en una extensión del estudio controlado a largo plazo. Los pacientes que recibieron ONGLYZA® durante las 24 semanas del periodo inicial del estudio mantuvieron la misma dosis de ONGLYZA® en la extensión a largo plazo. Todos los análisis de eficacia se basaron en datos obtenidos antes de la terapia de rescate. Todos los pacientes que recibieron placebo en el periodo de estudio inicial de 24 semanas y que no requirieron terapia de rescate hiperglucémica, recibieron 500 mg de metformina al entrar en el estudio de extensión a largo plazo. Para estos pacientes el cambio de HbA1c desde la línea basal fue +0.1% en la semana 102, mientras que el cambio desde la línea basal para los pacientes tratados con ONGLYZA®5 mg fue de -0.2%.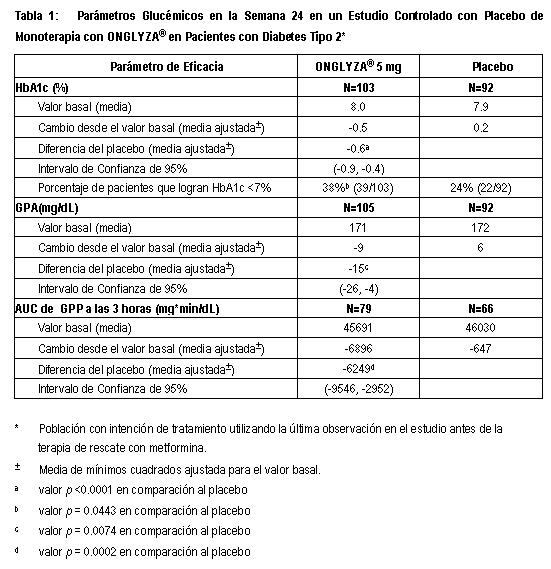
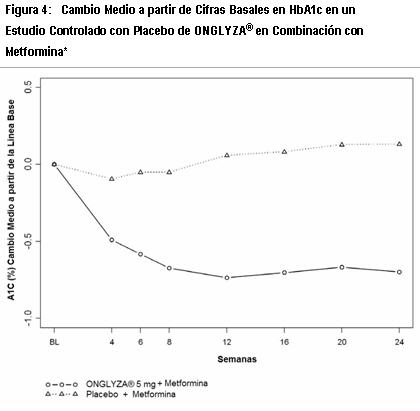
El segundo, fue un estudio de 24 semanas de monoterapia para evaluar un rango de regímenes de dosificación de ONGLYZA®. Los pacientes nunca antes tratados con diabetes inadecuadamente controlada (HbA1c ≥7% a ≤10%) se sometieron a un periodo inicial de 2 semanas, con ciego único, dieta, ejercicio y placebo. Un total de 365 pacientes fueron distribuidos aleatoriamente a recibir 2.5 mg cada mañana, 5 mg cada mañana, 2.5 mg con posible titulación hasta 5 mg cada mañana o 5 mg cada tarde de ONGLYZA® o placebo. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante los estudios fueron tratados con la terapia de rescate con metformina, adicionada a placebo o a ONGLYZA®. El número de pacientes distribuidos aleatoriamente por grupo de tratamiento estuvieron en el intervalo de 71 a 74. El tratamiento, ya sea con 5 mg de ONGLYZA® cada mañana o 5 mg cada tarde, proporcionó mejoras significativas en HbA1c versus placebo (-0.7% y -0.6%, respectivamente, en comparación con -0.3%). Mejora en el control glucémico - Terapia de combinación: Terapia de Combinación Adicionada a Metformina: Un total de 743 pacientes con diabetes tipo 2 participaron en este estudio controlado con placebo, doble ciego, aleatorizado, con duración de 24 semanas, para evaluar la eficacia y la seguridad de ONGLYZA® en combinación con metformina, en pacientes con control glucémico inadecuado (HbA1c ≥7% y ≤10%) con metformina sola. Se requirió que los pacientes estuvieran con una dosis estable de metformina (1500 mg a 2550 mg diariamente) durante al menos 8 semanas para ser reclutados en este estudio. Los pacientes que cumplieron los criterios de elegibilidad fueron reclutados en un periodo inicial de placebo con dieta y ejercicio, de dos semanas, con ciego único, durante el cual los pacientes recibieron metformina a su dosis de pre-estudio, hasta 2500 mg diariamente por la duración del estudio. Después del periodo inicial, los pacientes elegibles fueron distribuidos aleatoriamente a 2.5 mg, 5 mg ó 10 mg de ONGLYZA® o placebo además de su dosis actual de metformina de etiqueta abierta. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante el estudio fueron tratados con la terapia de rescate con pioglitazona, adicionada al placebo o a ONGLYZA® más metformina. Las titulaciones de dosis de ONGLYZA® y metformina no fueron permitidas en este estudio. En combinación con metformina, ONGLYZA® 5 mg proporcionó mejoras significativas en HbA1c, GPA y GPP en comparación con el grupo de placebo más metformina (Tabla 2). Se observaron reducciones en HbA1c en la semana 4 (Figura 4) y GPA en la semana 2 en el grupo de ONGLYZA® 5 mg más metformina en comparación al grupo de placebo más metformina, los puntos de evaluación mas tempranos. La proporción de pacientes que alcanzaron HbA1c < 7% (no obstante el valor basal) fue significativamente mayor en los grupos de tratamiento con ONGLYZA® 5 mg más metformina en comparación con el grupo del placebo más metformina. Se observaron reducciones significativas en el nivel de GPP a las 2 horas después de la prueba de tolerancia oral a la glucosa (COTG) estándar en el tratamiento con ONGLYZA® 5 mg más metformina (-58 mg/dL) en comparación con -18 mg/dL en el grupo de placebo más metformina. La proporción de pacientes que discontinuaron el tratamiento por falta de control glucémico o que fueron rescatados por cumplir los criterios glucémicos preespecificados, fue más alta en el grupo de placebo más metformina (27%) que en el grupo de ONGLYZA® 5 mg más metformina (13%). Valores basales mayores de HbA1c estuvieron asociados con un cambio promedio ajustado mayor, desde el basal, en HbA1c con ONGLYZA® 5 mg. El efecto de ONGLYZA® sobre los criterios de valoración de lípidos en este estudio fue similar al placebo. Se observaron reducciones similares en el peso corporal en pacientes que recibieron la terapia con ONGLYZA® y placebo (-0.9 kg y -0.9 kg, respectivamente).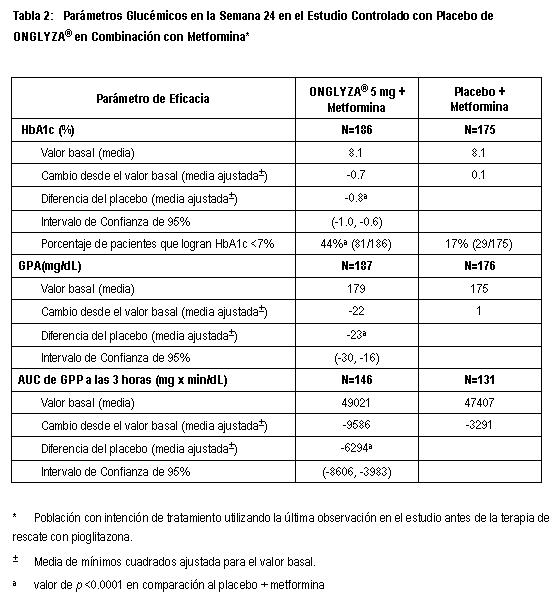
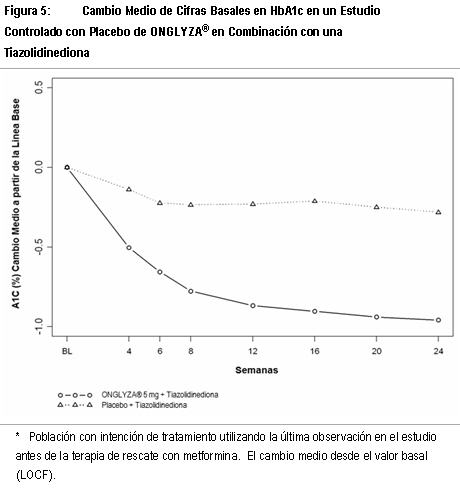
Estudio de Extensión Controlado a Largo Plazo: Los pacientes que fueron rescatados (basado en los niveles de glucosa predefinidos) durante las 24 semanas del periodo inicial del estudio, así como aquéllos que completaron todas las visitas durante las 24 semanas del periodo inicial del estudio sin necesidad de terapia de rescate fueron elegibles para entrar en un estudio controlado a largo plazo de extensión. Los pacientes que recibieron ONGLYZA® durante las 24 semanas del periodo inicial del estudio mantuvieron la misma dosis de ONGLYZA® en la extensión a largo plazo. Todos los análisis de eficacia se basaron en datos obtenidos antes de la terapia de rescate. El tratamiento con ONGLYZA® 5 mg más metformina estuvo asociado con una mayor reducción en HbA1c que en el grupo de placebo más metformina y el efecto relativo al placebo se mantuvo en la semana 102. El cambio de HbA1c para ONGLYZA® 5 mg más metformina comparado con el placebo más metformina fue -0.8% en la semana 102. Terapia de Combinación Adicionada a una Tiazolidinediona: Un total de 565 pacientes con diabetes tipo 2 participaron en este estudio controlado con placebo, doble ciego, aleatorizado, con duración de 24 semanas, para evaluar la eficacia y la seguridad de ONGLYZA® en combinación con una tiazolidinediona (TZD) en pacientes con control glucémico inadecuado (HbA1c ≥7% a ≤10.5%) sobre la TZD sola. Se requirió que los pacientes estuvieran en una dosis estable de pioglitazona (30 a 45 mg una vez al día) o rosiglitazona (4 mg una vez al día, u 8 mg, ya sea una vez al día o en dosis divididas de 4 mg) por al menos 12 semanas para ser reclutados en este estudio. Los pacientes que cumplieron los criterios de elegibilidad fueron reclutados en un periodo inicial con placebo, dieta y ejercicio, de dos semanas, con ciego único, durante el cual los pacientes recibieron TZD a su dosis pre-estudio por la duración del estudio. Después del periodo inicial, los pacientes elegibles fueron distribuidos aleatoriamente a 2.5 mg o 5 mg de ONGLYZA® o placebo, además de su dosis actual de TZD. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante el estudio fueron tratados con terapia de rescate con metformina, adicionada al placebo o a ONGLYZA® más TZD. La titulación de la dosis de ONGLYZA® o TZD no fue permitida durante el estudio. Un cambio en el régimen de TZD de rosiglitazona a pioglitazona a dosis terapéuticas equivalentes, específicas, fue permitido a discreción del investigador, si creía que era médicamente apropiado. En combinación con TZD, ONGLYZA® 5 mg proporcionó mejoras significativa en HbA1c, GPA y GPP en comparación con el grupo de tratamiento con placebo más TZD (Tabla 3). Se observaron reducciones en HbA1c (Figura 5) en la semana 4 y GPA en la semana 2 en el grupo de tratamiento con ONGLYZA® 5 mg más TZD con relación al grupo de placebo más TZD, en los puntos más tempranos de la evaluación. La proporción de los pacientes que logran HbA1c < 7% (no obstante el valor basal) fue significativamente mayor en el grupo de tratamiento con ONGLYZA® 5 mg más TZD en comparación con el grupo de placebo más TZD. Se observaron reducciones significativas en el nivel de GPP a las 2 horas después de la prueba de tolerancia oral a la glucosa (OGTT) estándar en el grupo de tratamiento con ONGLYZA® 5 mg más TZD (-65 mg/dL) en comparación con -15 mg/dL en el grupo de placebo más TZD. La proporción de pacientes que discontinuaron el tratamiento por falta de control glucémico o que fueron rescatados por cumplir los criterios glucémicos especificados fue de 10% en el grupo de placebo más TZD y 6% para el grupo de ONGLYZA® 5 mg más TZD. Valores basales mayores de HbA1c estuvieron asociados con un cambio medio ajustado mayor desde el basal, en HbA1c con ONGLYZA® 5 mg. El efecto de ONGLYZA® sobre los criterios de valoración de lípidos en este estudio fue similar al placebo. Se observaron incrementos pequeños en el peso corporal en los grupos de tratamiento ONGLYZA® 5 mg de y placebo (1.4 kg y 0.9 kg, respectivamente).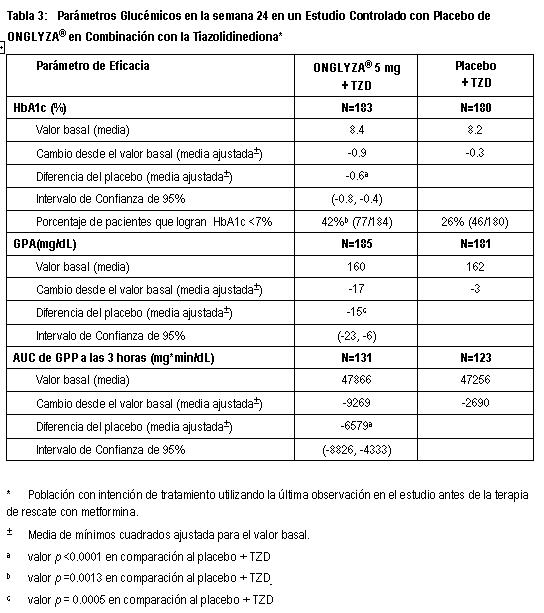
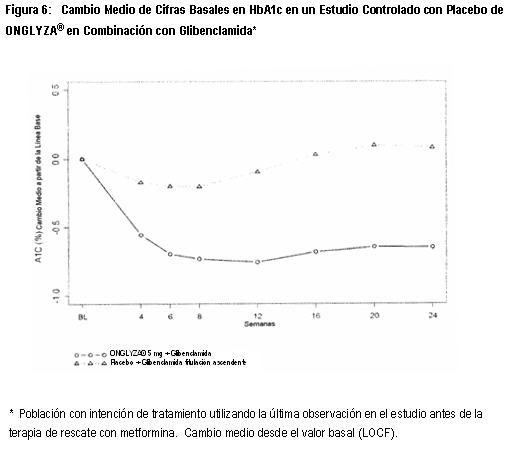
Estudio de Extensión Controlado a Largo Plazo: Los pacientes que fueron rescatados (basado en los niveles de glucosa predefinidos) durante las 24 semanas del periodo inicial del estudio, así como aquéllos que completaron todas las visitas durante las 24 semanas del periodo inicial del estudio sin necesidad de terapia de rescate fueron elegibles para entrar en un estudio controlado a largo plazo de extensión. Los pacientes que recibieron ONGLYZA® durante las 24 semanas del periodo inicial del estudio mantuvieron la misma dosis de ONGLYZA® en la extensión a largo plazo. Todos los análisis de eficacia se basaron en datos obtenidos antes de la terapia de rescate. El tratamiento con ONGLYZA® 5 mg más TZD estuvo asociado con una mayor reducción en HbA1c que en el grupo de placebo más TZD, y el efecto placebo se mantuvo hasta la Semana 76. El cambio de HbA1c para ONGLYZA® 5 mg más TZD comparado con placebo más TZD fue -0.9% en la Semana 76. Terapia de Combinación Adicionada a una Sulfonilurea: Un total de 768 pacientes con diabetes tipo 2 participaron en este estudio controlado con placebo, doble ciego, distribuidos aleatoriamente, con duración de 24 semanas, para evaluar la eficacia y la seguridad de ONGLYZA® en combinación con sulfonilurea (SU) en pacientes con control glucémico inadecuado al momento del enrolamiento (HbA1c ≥7.5% a ≤10%) en una dosis submáxima de SU sola. Se requirió que los pacientes estuvieran en una dosis submáxima de SU por 2 meses o más tiempo para ser reclutados en este estudio. En este estudio, ONGLYZA® en combinación con una dosis intermedia fija de SU fue comparada con la titulación a dosis más altas de SU. Los pacientes que cumplieron los criterios de elegibilidad fueron reclutados en un periodo inicial de dieta y ejercicio de 4 semanas, con ciego único y se les administró glibenclamida 7.5 mg una vez al día. Después del periodo inicial, los pacientes elegibles con HbA1c ≥7% a ≤10% fueron distribuidos aleatoriamente ya sea a 2.5 mg ó 5 mg de ONGLYZA® más 7.5 mg de glibenclamida, o placebo más una dosis diaria total de 10 mg de glibenclamida. Los pacientes que recibieron placebo fueron elegibles para tener una titulación ascendente de glibenclamida hasta una dosis diaria total de 15 mg. La titulación ascendente de glibenclamida no fue permitida en pacientes que recibieron ONGLYZA® a 2.5 ó 5 mg. La glibenclamida podía ser titulada en forma descendente, una sóla vez, en cualquier grupo de tratamiento, durante el periodo de estudio de 24 semanas, debido a hipoglucemia, si lo consideraba necesario el investigador. Aproximadamente a 92% de los pacientes en el grupo de placebo más glibenclamida, ésta se les tituló en forma ascendente a una dosis diaria total final de 15 mg durante el periodo del estudio. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante el estudio, fueron tratados con metformina de rescate, adicionada al grupo de ONGLYZA® más glibenclamida o al grupo de placebo más glibenclamida con titulación ascendente. La titulación de la dosis de ONGLYZA® no fue permitida durante el estudio. En combinación con glibenclamida, ONGLYZA® 5 mg, proporcionó mejoras significativas enHbA1c, GPA y GPP en comparación con el grupo de placebo más glibenclamida con titulación ascendente (Tabla 4). En las etapas tempranas de evaluación, se observaron reducciones en HbA1c (Figura 6) en la semana 4 y GPA en la semana 2 en el grupo de tratamiento con ONGLYZA® 5 mg más glibenclamida, con relación al grupo de placebo más glibenclamida con titulación ascendente. La proporción de los pacientes que alcanzan HbA1c < 7% (no obstante del valor basal) fue significativamente mayor en el grupo de tratamiento con ONGLYZA® 5 mg más glibenclamida, en comparación con el grupo de placebo más glibenclamida con titulación ascendente. Se observaron reducciones significativas en el nivel de GPP de 2 horas después de la prueba de tolerancia oral a la glucosa (COTG), estándar, en el grupo de tratamiento con 5 mg de ONGLYZA® más glibenclamida (-34 mg/dL) en comparación con 8 mg/dL en el grupo de placebo más glibenclamida con titulación ascendente. La proporción de pacientes que discontinuaron el tratamiento por falta de control glucémico o que fueron rescatados para cumplir los criterios glucémicos especificados previamente, fue más alta en el grupo de placebo más glibenclamida con titulación ascendente (30%) que en el grupo de ONGLYZA® 5 mg más glibenclamida (17%). Valores basales mayores de HbA1c estuvieron asociados con un cambio medio ajustado mayor desde el basal en HbA1c con ONGLYZA® 5 mg. El efecto de ONGLYZA® sobre los criterios de valoración lipídicos en este estudio fue similar al placebo. En este estudio se observaron incrementos pequeños en el peso corporal en pacientes tratados con 5 mg de ONGLYZA® más glibenclamida y con placebo más glibenclamida con titulación ascendente (0.8 kg versus 0.3 kg, p=0.012).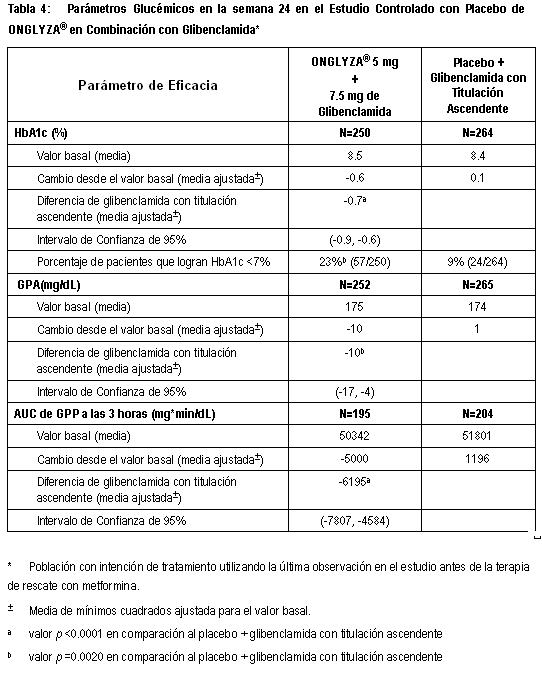
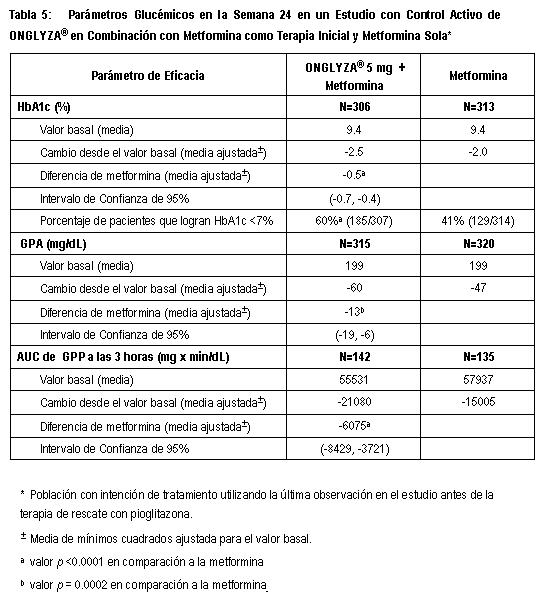
Estudio de Extensión Controlado a Largo Plazo: Los pacientes que fueron rescatados (basado en los niveles de glucosa predefinidos) durante las 24 semanas del periodo inicial del estudio, así como aquéllos que completaron todas las visitas durante las 24 semanas del periodo inicial del estudio sin necesidad de terapia de rescate, fueron elegibles para entrar en un estudio de extensión, controlado a largo plazo. Los pacientes que recibieron ONGLYZA® durante las 24 semanas del periodo inicial del estudio mantuvieron la misma dosis de ONGLYZA® en la extensión a largo plazo. Los pacientes que recibieron placebo y que participaron en la extensión a largo plazo sin recate fueron elegibles para una dosis cegada de glibenclamida con titulación ascendente hasta una dosis total diaria de 20 mg. Todos los análisis de eficacia se basaron en datos obtenidos antes de la terapia de rescate. El tratamiento con ONGLYZA® 5 mg más glibenclamida estuvo asociado con una mayor reducción en HbA1c que en el grupo de placebo más glibenclamida con titulación ascendente y el efecto relativo al placebo se mantuvo en la Semana 76. El cambio de HbA1c para ONGLYZA® 5 mg más glibenclamida comparado con el placebo más glibenclamida titulación ascendente fue -0.7% en la Semana 76. Combinación con Metformina como Terapia Inicial: Un total de 1306 pacientes con diabetes tipo 2 nunca antes tratados, participaron en este estudio aleatorizado, doble ciego, con control activo y 24 semanas de duración, para evaluar la eficacia y seguridad de ONGLYZA® como terapia de combinación inicial con metformina en pacientes con control glucémico inadecuado (HbA1c ≥8% a ≤12%) con dieta y ejercicio únicamente. Se requirió que los pacientes no hubieran recibido tratamiento previo para ser reclutados en este estudio. Los pacientes que cumplieron el criterio de elegibilidad fueron reclutados con un periodo de inicio de 1 semana con ciego único, recibiendo placebo, dieta y ejercicio. Los pacientes fueron distribuidos aleatoriamente a uno de los cuatro grupos de tratamiento: ONGLYZA® 5 mg + metformina 500 mg, saxagliptina 10 mg + metformina 500 mg, saxagliptina 10 mg + placebo o metformina 500 mg + placebo. ONGLYZA® fue dosificado una vez al día. Durante las semanas 1 a la 5, en los grupos de ONGLYZA® 5 mg y saxagliptina 10 mg más metformina, y en el grupo de metformina sola, metformina fue titulada en forma ascendente con base en los niveles de GPA con incrementos de 500 mg por día según lo tolerado hasta un máximo de 2000 mg diarios. Los pacientes que fallaron en cumplir las metas glucémicas específicas durante el estudio, fueron tratados con terapia de rescate con pioglitazona adicionada a la terapia recibida. La terapia inicial con la combinación de ONGLYZA® 5 mg más metformina proporcionó disminución significativa en HbA1c, GPA y GPP en comparación con metformina so
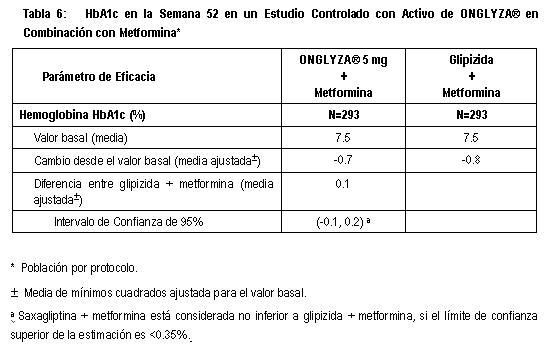
Estudio de Extensión Controlado a Largo Plazo: Los pacientes que fueron rescatados (basados en los niveles de glucosa predefinidos) durante las 24 semanas del periodo inicial del estudio, así como aquéllos que completaron todas las visitas durante las 24 semanas del periodo inicial del estudio sin necesidad de terapia de rescate, fueron elegibles para entrar en un estudio de extensión controlado a largo plazo. Los pacientes que recibieron ONGLYZA® durante las 24 semanas del periodo inicial del estudio, mantuvieron la misma dosis de ONGLYZA® en la extensión a largo plazo. Todos los análisis de eficacia se basaron en datos obtenidos antes de la terapia de rescate. El tratamiento con 5 mg de ONGLYZA® más metformina estuvo asociado con una mayor reducción en HbA1c que en el grupo de placebo más metformina y el efecto de metformina se mantuvo a la Semana 76. El cambio de HbA1c para 5 mg de ONGLYZA®más metformina comparado con placebo más metformina fue -0.5% en la Semana 76. Terapia de combinación adicionada a metformina en comparación con glipizida en combinación adicionada a metformina: Un total de 858 pacientes con diabetes tipo 2, participaron en este estudio controlado con activo, doble ciego, distribuidos aleatoriamente, con duración de 52 semanas, para evaluar la eficacia y la seguridad de ONGLYZA® en combinación con metformina comparada con una sulfonilurea (SU) en combinación con metformina, en pacientes con control glucémico inadecuado con metformina sola (HbA1c > 6.5% y ≤10%). Se requirió que los pacientes estuvieran en una dosis estable de metformina (por lo menos 1500 mg) por lo menos 8 semanas antes de ser enrolados en el estudio. Los pacientes que cumplieron con los criterios de elegibilidad fueron enrolados en un estudio doble ciego, con dieta y ejercicio y un periodo de introducción de dos semanas con placebo, durante el cual los pacientes recibieron metformina (1500 a 3000 mg en función de su dosis previa al estudio) para la duración del estudio. Tras el período de introducción, los pacientes elegibles fueron aleatorizados para ONGLYZA® 5 mg o glipizida 5 mg, además de su dosis actual de metformina en el periodo abierto. El grupo de pacientes a los que se les administró glipizida más metformina, fueron titulados durante las primeras 18 semanas hasta un efecto óptimo (GPA ≤6.1 mmol/L, ≤110 mg/dL) o a la dosis máxima tolerable, utilizando una técnica de doble simulación hasta un máximo de 20 mg por día (dosis media de 15 mg). De acuerdo al análisis primario del protocolo establecido, la reducción de HbA1c para ONGLYZA® 5 mg adicionada a metformina no fue inferior a glipizida adicionada a metformina (Tabla 6). El análisis por intención a tratamiento mostró resultados consistentes. Los eventos de hipoglucemia en pacientes tratados con ONGLYZA® 5mg se presentaron en una proporción significativamente más baja, 3% (19 eventos en 13 pacientes) en comparación a glipizida, 36.3% (750 eventos en 156 pacientes). Los pacientes tratados con ONGLYZA® 5 mg mostraron una disminución significativa del peso corporal desde el valor basal, en comparación con la ganancia de peso de los tratados con glipizida (-1.1 Kg comparado con +1.1Kg, p < 0.0001).
Estudio de extensión controlado a largo plazo: Los pacientes que completaron el periodo inicial de 52 semanas del estudio fueron elegibles para entrar en el estudio de extensión controlado a largo plazo de 52 semanas. Los pacientes mantuvieron la misma dosis de Onglyza 5 mg o glipizida en la extensión a largo plazo. Los cambios en los valores basales de HbA1c fueron -0.4% para Onglyza 5 mg (N=184) adicionada a metformina y -0.3% para glipizida (N=160) adicionada a metformina a la semana 104. El tratamiento con Onglyza 5 mg más metformina resultó en una menor proporción de pacientes con eventos hipoglucémicos, 3.5 % (24 eventos en 15 pacientes) versus 38.4% (896 eventos para 165 pacientes) para el tratamiento con glipizida más metformina. El tratamiento con Onglyza 5 mg más metformina produjo una reducción en el peso medio corporal en comparación con los valores basales (-1.5 Kg) mientras que el tratamiento con glipizida más metformina produjo un incremento en el peso corporal medio comparado con los valores basales (+1.3 Kg) a la semana 104. El perfil de seguridad en general de Onglyza versus glipizida en el periodo del tratamiento a largo plazo fue consistente con la observada previamente en el periodo inicial del tratamiento en 52 semanas. Terapia de Combinación Adicionada con Insulina (con o sin metformina): Un total de 455 pacientes con diabetes tipo 2 participaron en el estudio controlado con placebo, doble ciego, distribuidos aleatoriamente, de 24 semanas de duración, para evaluar eficacia y seguridad de ONGLYZA® en combinación con insulina en pacientes con control glucémico inadecuado (HbA1c ≥7.5% y ≤11%), con insulina sola (N=141) o insulina en combinación con una dosis estable de metformina (N=314). Se requirió que los pacientes estuvieran con una dosis estable de insulina (≥ 30 unidades a ≤ 150 unidades, diariamente) con una variación ≤ 20% en la dosis total diaria durante ≥ 8 semanas previo a la selección con o sin metformina. Los pacientes estuvieron con insulina de acción intermedia o prolongada (basal) o insulina premezclada. Los pacientes que utilizaban insulina de acción corta fueron excluidos a menos que ésta fuera administrada como parte de una insulina premezclada. Los pacientes que cumplieron con los criterios de elegibilidad fueron enrolados en un periodo de inducción con placebo, ciego simple, de cuatro semanas, con dieta y ejercicio durante el cual los pacientes recibieron insulina (y metformina, si aplicaba) en la dosis pre-estudio. Después del periodo de inducción, los pacientes seleccionados fueron aleatorizados con 5 mg de ONGLYZA® o placebo en adición para continuar con su dosis habitual de insulina (y metformina, si aplica). Los pacientes mantuvieron una dosis estable de insulina cuando fue posible. Los pacientes que fallaron el cumplimiento de las metas glucémicas o quienes incrementaron su dosis de insulina por ≥ 20%, fueron rescatados y posteriormente cambiados a un régimen de dosis de insulina mas flexible. No se permitió la titulación de la dosis de ONGLYZA® y metformina. ONGLYZA® 5 mg adicionada a insulina, con o sin metfomina, proporciona disminuciones significativas en HbA1c y GPA en comparación con placebo adicionado a insulina, con o sin metformina (Tabla 7). Se alcanzaron reducciones de HbA1c similares contra placebo, en pacientes que utilizan ONGLYZA® 5 mg adicionada a insulina sola y ONGLYZA® adicionada a insulina en combinación con metformina (-0.4% y -0.4%, respectivamente). La proporción de pacientes que fueron suspendidos por falta de control glucémico o rescatados fue de 23% en el grupo de ONGLYZA® 5 mg adicionada a insulina y 32% en el grupo de placebo adicionado a insulina.
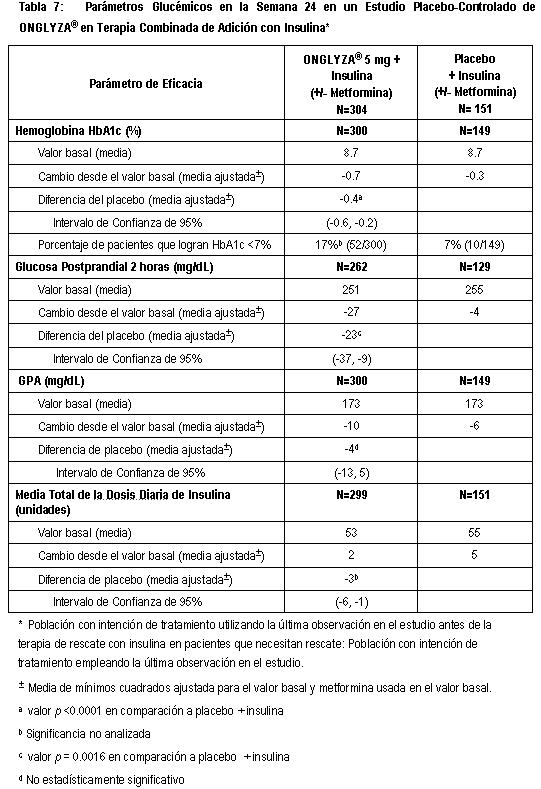
Estudio de extensión controlado a largo plazo: Los pacientes que completaron todas las visitas durante el periodo inicial de 24 semanas del estudio fueron elegibles para entrar al estudio de extensión controlado a largo plazo. Los pacientes que recibieron ONGLYZA en el periodo inicial de 24 semanas mantuvieron la misma dosis de ONGLYZA en el periodo de extensión a largo plazo, pero los pacientes que completaron el periodo de 24 semanas del estudio con una dosis estable de insulina cambiaron a un régimen de dosis flexible de insulina durante el periodo de extensión. Todos los análisis de eficacia se basaron en los datos independientemente de la dosis de insulina. El tratamiento con ONGLYZA 5 mg adicionada a insulina con o sin metformina se asoció a una mayor reducción de niveles de HbA1c que el uso de placebo adicionado a insulina con o sin metformina y el efecto relacionado al placebo fue sostenido hasta la semana 52. El cambio de niveles de HbA1c para saxagliptina 5 mg más insulina (N=244) en comparación con el placebo más insulina (N=124) fue -0.4% a la semana 52. Terapia de adición combinada con metformina más sulfonilurea. Un total de 257 pacientes con diabetes tipo 2 participaron en el estudio placebo-controlado, doble ciego, con distribución aleatorizada, de 24 semanas, para evaluar la eficacia y seguridad de ONGLYZA en combinación con metformina más sulfonilurea en pacientes con control glucémico inadecuado (HbA1c ≥ 7% y ≤ 10%). Se requirió que los pacientes estuvieran con una dosis combinada estable de metformina de liberación prolongada o de liberación inmediata (en dosis máxima tolerada, con una dosis mínima para ser enrolados de 1500 mg) y sulfonilurea (en dosis máxima tolerada, con dosis mínima para ser enrolados ≥ 50% de la dosis máxima recomendada) por al menos 8 semanas antes del enrolamiento. Los pacientes que cumplieron con los criterios de elegibilidad fueron ingresados en un periodo de enrolamiento de 2 semanas para permitir la evaluación de los criterios de inclusión/exclusión. Después del periodo de enrolamiento, los pacientes seleccionados fueron aleatorizados a doble ciego con ONGLYZA (5 mg una vez al día) o doble ciego con placebo durante 24 semanas. Durante el periodo de tratamiento de 24 semanas, los pacientes debían recibir metformina y sulfonilurea a la misma dosis constante observada en el momento de enrolamiento. La sulfonilurea podría ser titulada una vez a la baja en el caso de un evento hipoglucémico o eventos recurrentes hipoglucémicos menores. Durante el periodo del tratamiento no se permitió la titulación (a la alza o a la baja) de los medicamentos. ONGLYZA en combinación con metformina más sulfonilurea proporcionó mejoras significativas en los niveles de HbA1c y GPP en comparación con placebo en combinación con metformina más una sulfonilurea (Tabla 8).
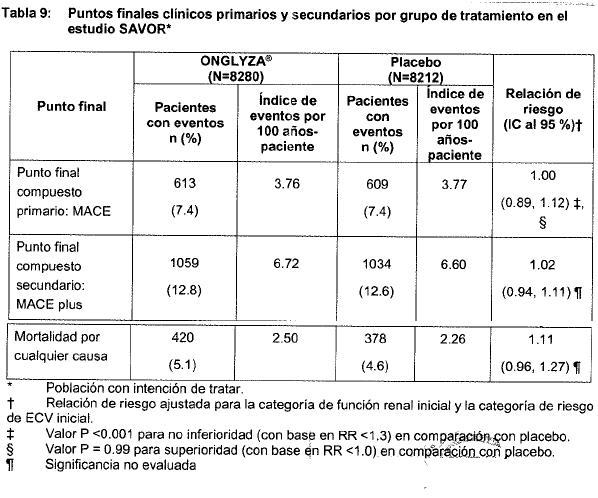
Poblaciones Especiales: Uso en insuficiencia renal: Un total de 170 pacientes participaron en un estudio controlado con placebo, doble ciego, aleatorizado, con duración de 12 semanas, para evaluar la eficacia y la seguridad de saxagliptina 2.5 mg una vez al día en comparación con pacientes diabéticos tipo 2 y con daño renal (moderado, severo, ESRD) tratados con placebo. En este estudio el 98.2% de los pacientes que entraron y continuaron con medicación antidiabética (insulina y/o medicamento antidiabético oral) además del fármaco en estudio (75.3% con insulina y 31.2% con el medicamento antidiabético oral; algunos recibieron ambos). El tratamiento con 2.5 mg de saxagliptina proporcionó mejoras significativas en HbA1c en comparación con placebo (la reducción media desde el valor basal en la semana 12, para el grupo de saxagliptina fue de -0.9% y para el grupo placebo -0.4%, p = 0.007). El perfil de seguridad de saxagliptina en este estudio fue consistente con lo observado en la experiencia de estudios clínicos previos. No se observaron efectos adversos sobre la función renal. El número de sujetos con cualquier caso de hipoglucemia fue similar entre los grupos de tratamiento. Estudio de extensión controlado a largo plazo: Los pacientes que completaron el periodo inicial de estudio a 12 semanas, fueron elegibles para entrar a la extensión del estudio controlado a largo plazo de 40 semanas. Los pacientes que recibieron saxagliptina en el periodo de estudio inicial de 12 semanas, mantuvieron la misma dosis de saxagliptina en la extensión a largo plazo. La mayoría de los sujetos (76%) en ambos grupos de tratamiento que recibieron insulina u otros medicamentos antihiperglucémicos no tuvieron cambio en la dosis o tipo de medicamento durante el periodo de tratamiento de 52 semanas. En sujetos en los que hubo un cambio significativo en el tipo o dosis de insulina (mayor de ±20%) o medicamentos antihiperglucémicos, a partir de esto los resultados de eficacia fueron excluidos del análisis. El tratamiento con saxagliptina 2.5 mg proporcionó una mejora significativa en HbA1c versus placebo (reducción media a partir de la línea base de -1.4 % en la semana 52 para el grupo de saxagliptina y -0.5% para el grupo de placebo). El perfil de seguridad de saxagliptina en el periodo de tratamiento a largo plazo fue consistente con el observado previamente en la experiencia de estudios clínicos y el observado en el periodo de tratamiento a corto plazo (12 semanas). Uso geriátrico: De los 16,492 pacientes distribuidos aleatoriamente en el estudio SAVOR, 8561 pacientes (51.9%) tenían 65 años o más y 2330 (14.1%) tenían 75 años o más. El número de pacientes de 65 años o más tratados con ONGLYZA en el estudio SAVOR fue 4290, mientras que el número de pacientes mayores de 75 años fue de 1169. Del número total de sujetos (N=4148) en seis estudios doble ciego, controlados, sobre la seguridad y eficacia clínica de ONGLYZA, 634 (15.3%) pacientes tenían una edad de 65 años o más, de los cuales 59 (1.4%) pacientes tenían 75 años y más. No se observaron diferencias generales en la seguridad o efectividad entre los sujetos mayores de 65 años y de 75 años y entre sujetos más jóvenes. Seguridad cardiovascular: En el estudio de evaluación de desenlaces cardiovasculares de saxagliptina registrados en pacientes con diabetes mellitus-trombólisis en infarto de miocardio (Saxagliptin Assesment of Vascular Outcomes Recorder in Patients with Diabetes Mellitus-thrombolysis in Miocardial Infarction, SAVOR por sus siglas en inglés) se evaluó el efecto de saxagliptina en la incidencia de eventos cardiovasculares (ECV) en 16,492 pacientes adultos con diabetes tipo 2 quienes ya tenian ECV o presentaban múltiples factores de riesgo para enfermedad vascular, incluyendo a pacientes con insuficiencia renal moderada o grave. Se reclutaron pacientes ≥40 años de edad, diagnosticados con diabetes tipo 2 y con HbA1C ≥6.5%, y con ECV declarada o múltiples factores de riesgo CV. Los pacientes fueron asignados aleatoriamente al grupo de placebo (n=8212) o de saxagliptina (5 mg ó 2.5 mg para pacientes con insuficiencia renal moderada o grave) una vez al día (n=8280). La asignación aleatoria a los grupos de saxagliptina y placebo se estratificó por riesgo CV con 3533 pacientes (21.4 %) únicmaente con factores de riesgo CV y 12,959 pacientes (78.6%) con ECV declarada y por insuficiencia renal que incluyó 13,916 pacientes (84.4%), con función renal normal o insuficiencia leve, 2240 pacientes (13.6%) con insuficiencia moderada y 336 pacientes (2.0%) con insuficiencia renal grave. Los pacientes con ECV declarada se definieron por medio de antecedentes de cardiopatía isquémica, enfermedad vascular periférica, o accidente vascular cerebral isquémico. Para los pacientes con factores de riesgo CV sólo se tomó la edad (hombres ≥55 años y mujeres ≥60 años) como factor de riesgo y al menos un factor de riesgo adicional de dislipidemia, hipertensión o tabaquismo actual. Las características demográficas y basales en los pacientes fueron equilibradas entre los grupos de saxagliptina y placebo. La población del estudio comprendió 67% de hombres y 33% de mujeres con una edad media de 65 años en la aleatorización. De los 16,492 pacientes asignados al azar, 8561 pacientes (52%) tenía 65 años o más y 2330 (14%) tenía 75 años o más. Todos los pacientes del estudio tenían DMT2 con una duración media de 12 años (mediana = 10.3) y un nivel de HbA1C medio de 8.0% (mediana = 7.6%). En general, 25% de los pacientes tenía niveles iniciales de HbA1C < 7%. Se les dio seguimiento a los pacientes por una duración media de 2 años (mediana = 2.0). El uso de medicamentos concomitantes fue similar para los dos grupos de tratamiento. En general, el uso de medicamentos para tratar la diabetes concordó con la práctica local de tratamiento y el programa clínico de saxagliptina (metformina 69%, insulina 41%, sulfonilureas 40% y TZD 6%). El uso de medicamentos para la ECV también concordó con la práctica local de tratamiento (inhibidores de la ECA o BRA 79%, estatinas 78%, aspirina 75%, betabloqueadores 62% y medicamentos antiplaquetarios diferentes a la aspirina 24%). Se trató aproximadamente a 6% de los pacientes únicamente con dieta y ejercicio al inicio. Los medicamentos concomitantes se administraron durante el estudio conforme a los objetivos de los lineamientos locales para control glucémico y reducción de riesgo CV para minimizar las diferencias entre los dos grupos de tratamiento, especialmente para control glucémico. El punto final primario de seguridad y eficacia fue un punto final compuesto, conformado por el tiempo hasta la primera incidencia de cualquiera de los siguientes eventos adversos CV graves (MACE por sus siglas en inglés): muerte CV, infarto de miocardio no letal o accidente vascular cerebral isquémico no letal. El objetivo final primario de seguridad de este estudio fue establecer que el límite superior del IC al 95% bilateral para la relación de riesgo calculada, que compara la incidencia del punto final compuesto de muerte CV, IM no letal o accidente vascular cerebral isquémico no letal observado con saxagliptina en comparación con el observado en el grupo de placebo fue de < 1.3. El objetivo final primario de eficacia fue determinar, como una evaluación de superioridad, si el tratamiento con saxagliptina, en comparación con placebo cuando se añadió a la terapia de base actual, resultaba en una reducción significativa en el punto final primario de MACE. El primer punto final secundario de eficacia fue un punto final compuesto conformado por el tiempo hasta la incidencia de MACE más hospitalización por insuficiencia cardiaca, hospitalización por angina de pecho inestable u hospitalización por revascularización coronaria (MACE plus). El siguiente punto final secundario de eficacia fue determinar si el tratamiento con saxagliptina en comparación con placebo cuando se añadió a la terapia de base actual en pacientes con DMT2 provocaría una reducción de mortalidad por cualquier causa. Se evaluó la seguridad cardiovascular de saxagliptina en el estudio SAVOR, donde se estableció que saxagliptina no incrementó el riesgo de ECV (muerte CV, infarto de miocardio no letal o accidente vascular cerebral isquémico no letal) en pacientes con DMT2 en comparación con placebo cuando se añadió a la terapia de base actual (RR 1.00; IC al 95%: 0.89, 1.12; P < 0.001 para no inferioridad). El punto final primario de eficacia no demostró una diferencia estadísticamente significativa en eventos adversos coronarios graves para saxagliptina en comparación con placebo cuando se añadió a la terapia de base actual en pacientes con DMT2.
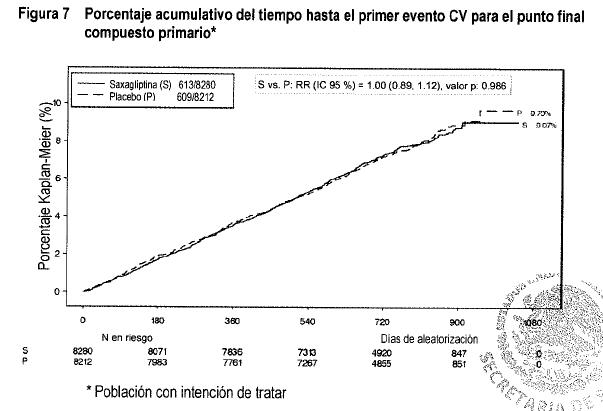
Los eventos se acumularon continuamente con el paso del tiempo, y las tasas de eventos para ONGLYZA y placebo no discrepan de forma notable con el paso del tiempo. Un componente del punto final compuesto secundario, hospitalización por insuficiencia cardiaca, presentó mayor incidencia en el grupo de saxagliptina (3.5%) en comparación con el grupo de placebo (2.8%) con una significativa estadística nominal (es decir, sin ajuste para pruebas o puntos finales múltiples) que favorece al grupo de placebo [RR = 1.27; (IC al 95% 1.07, 1.51); P = 0.007]. Definitivamente no se pudieron identificar los factores clínicamente relevantes predictivos del incremento de riesgo relativo en el tratamiento con saxagliptina. Los pacientes con mayor riesgo de hospitalización por insuficiencia cardiaca, independientemente del tratamiento asignado, se pudieron identificar por medio de factores de riesgo conocidos para insuficiencia cardiaca tales como antecedentes iniciales de insuficiencia cardiaca o disfunción renal. Sin embargo, los pacientes en tratamiento con saxagliptina, con antecedentes de insuficiencia cardiaca o disfunción renal al inicio, no tuvieron incremento de riesgo relativo a la administración de placebo para los puntos finales compuestos primarios o secundarios o mortalidad por cualquier causa. No se observó riesgo incrementado para el punto final primario entre saxagliptina y placebo en ninguno de los siguientes subgrupos: ECV, múltiples factores de riesgo por ECV, insuficiencia renal leve, moderada o grave, edad, sexo, raza, región, duración de la diabetes tipo 2, antecedentes de insuficiencia cardiaca, HbA1C inicial, proporción de albumina/creatinina, medicamento antidiabético al inicio, o uso al inicio de estatinas, aspirina, inhibidores de la ECA, BRA, betabloqueadores, o medicamentos antiplaquetarios. A pesar de la administración activa de la terapia antidiabética concomitante en ambos grupos de estudio, los niveles medios de HbA1C fueron menores en el grupo de saxagliptina en comparación con el grupo de placebo en el Año 1 (7.6% frente a 7.9%, a diferencia de -0.35% [IC al 95%: -0.38, -0.31]) y al Año 2 (7.6% frente al 7.9%, diferencia de -0.30% [IC al 95%: -o.34, -0.26]). Las proporciones de pacientes con HbA1C < 7% en el grupo de saxagliptina en comparación con el grupo de placebo fueron de 38% frente a 27% en el Año 1 y 38% frente a 29% en el Año 2. En comparación con placebo, saxagliptina resultó en una necesidad menor de iniciar con un nuevo medicamento oral para la diabetes, insulina o incrementar la dosis actual. La mejoría en HbA1C y la proporción de pacientes que alcanzaron las metas de HbA1C, entre los pacientes tratados con saxagliptina, fueron obtenidas a pesar de las menores tasas de ajustes ascendentes en medicamentos contra la diabetes o el inicio de nuevos medicamentos contra la diabetes o insulina, en comparación con placebo.
Contraindicaciones: ONGLYZA® está contraindicada en pacientes con una historia de cualquier reacción seria de hipersensibilidad, tal como anafilaxia o angioedema con cualquier inhibidor de DPP4.
Precauciones generales: Uso en pacientes con insuficiencia renal: En pacientes con TFG estimada < 45mL/min/1.73m2, la dosis es 2.5mg una vez al día (Ver Dosis y vía de administración). La evaluación de la función renal es recomendada antes del inicio del tratamiento con ONGLYZA® y posteriormente de una manera periódica. Uso con medicamentos que se conoce provocan hipoglucemia: Se sabe que los agentes antihiperglucémicos de la clase de la sulfonilurea y la insulina provocan hipoglucemia. Por tanto, puede ser requerida una dosis menor de sulfonilurea o insulina para reducir el riesgo de hipoglucemia cuando se utiliza en combinación con ONGLYZA® (Ver Reacciones secundarias y adversas). Reacciones de Hipersensibilidad: Durante la experiencia postcomercialización, las siguientes reacciones adversas se han reportado con el uso de saxagliptina: reacciones de hipersensibilidad serias, incluyendo anafilaxia y angioedema. Puesto que estas reacciones se han reportado voluntariamente de una población de tamaño incierto, no es posible estimar su frecuencia de manera confiable. Si se sospecha de alguna reacción seria de hipersensibilidad a saxagliptina, suspenda ONGLYZA®, valore otras causas potenciales del evento, y determine un tratamiento alternativo para diabetes (Ver Contraindicaciones). Pancreatitis: Durante la experiencia post-comercialización, se han recibido reportes de eventos adversos espontáneos de pancreatitis aguda. Los pacientes deben ser informados de los síntomas característicos de la pancreatitis aguda: dolor abdominal severo y persistente. Si se sospecha de pancreatitis, el uso de ONGLYZA® debe ser suspendido (Véase Reacciones secundarias y adversas). En la evaluación de resultados vasculares registrados en pacientes con diabetes mellitus-trombólisis en infarto de miocardio, estudio SAVOR, por sus siglas en inglés (Saxagliptin Assesment of Vascular Outcomes Recorder), la incidencia de eventos de pancreatitis relacionados fue de 0.3%, tanto en pacientes tratados con ONGLYZA® como en pacientes tratados con placebo en la población con intención de tratamiento (Ver Reacciones Secundarias y Adversas). Uso pediátrico: La seguridad y la efectividad de ONGLYZA® en pacientes pediátricos y adolescentes menores de 18 años no ha sido todavía establecida por lo que no se recomienda. Uso geriátrico: Saxagliptina y su metabolito principal son eliminados en parte por el riñón. Debido a que es más probable que los pacientes de edad avanzada tengan función renal disminuida, se debe tener cuidado en la selección de la dosis en los sujetos de edad avanzada con base en la función renal (Ver Dosis y vía de administración y Farmacocinética y Farmacodinamia). Insuficiencia cardiaca: En el estudio SAVOR, se observó un aumento en el índice de hospitalización por insuficiencia cardiaca en los pacientes tratados con saxagliptina en comparación con el placebo, aunque no se ha establecido una relación causal. Se justifica la precaución si ONGLYZA® se usa en pacientes que tienen factores de riesgo conocidos de hospitalización por causa de insuficiencia cardiaca, tales como antecedentes de insuficiencia cardiaca o insuficiencia renal de moderada a grave. Se debe advertir a los pacientes sobre los síntomas característicos de insuficiencia cardiaca y reportar de inmediato dichos síntomas (Ver Farmacocinética y Farmacodinamia). Artralgia: En informes posteriores a la comercialización se ha reportado dolor articular con inhibidores DPP4, el cual puede ser severo. Los pacientes sienten alivio de los síntomas después de suspender el medicamento y algunos experimentaron la reaparición de los síntomas con la reintroducción del mismo u otro inhibidor DPP4. La aparición de síntomas después de iniciar con la terapia del medicamento puede ser rápida o puede ocurrir después de largos periodos del tratamiento. Si un paciente presenta dolor articular severo, la continuación de la terapia con el medicamento debe evaluarse de manera individual. (Ver Reacciones Secundarias y Adversas). Efectos sobre la habilidad para conducir y para usar maquinaria: No se han realizado estudios sobre los efectos en la habilidad para conducir y utilizar maquinaria. Sin embargo, cuando se conducen y se operan máquinas, se debe tener en cuenta que se ha reportado vértigo con saxagliptina.
Restricciones de uso durante el embarazo y la lactancia: No existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción animal no siempre predicen la respuesta en humanos, ONGLYZA® debe ser utilizada durante el embarazo únicamente si es claramente necesario, valorando riesgo-beneficio. Saxagliptina cruza la placenta hacia el feto después de la administración en ratas preñadas. Saxagliptina es secretada en la leche de ratas lactantes. No se sabe si saxagliptina es secretada en la leche humana. Debido a que muchos fármacos son secretados en la leche humana, se debe tener precaución cuando ONGLYZA® sea administrada a una mujer que esté lactando.
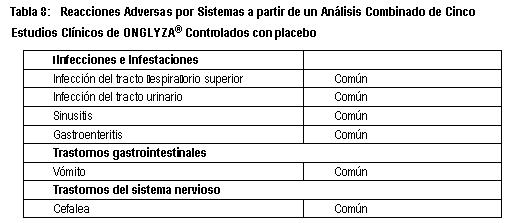
Reacciones secundarias y adversas: Experiencia clínica: Más de 17,000 pacientes con diabetes tipo 2 han sido tratados con ONGLYZA® en estudios clínicos aleatorizados, controlados y doble ciego. Reacciones adversas relacionadas con ONGLYZA® en el estudio SAVOR: El estudio SAVOR incluyó a 8240 pacientes tratados con ONGLYZA® 5 mg ó 2.5 mg una vez al día y 8173 pacientes con placebo. La duración media de exposición a ONGLYZA® independientemente de las interrupciones, fue de 1.8 años. Un total de 3698 pacientes (45%) fueron tratados con ONGLYZA® durante 2 y 3 años. La incidencia total de eventos adversos en pacientes tratados con ONGLYZA en este estudio fue similar con placebo (72.5% frente a 72.2%, respectivamente). La suspensión de la terapia debido a eventos adversos fue similar entre los dos grupos de tratamiento (4.9% en el grupo de ONGLYZA y 5.0% en el grupo de placebo). Se evaluó la seguridad cardiovascular de saxagliptina en el estudio SAVOR, donde se estableció que saxagliptina no incrementó el riesgo cardiovascular (muerte CV, IM no letal o accidente vascular cerebral isquémico no letal) en pacientes con diabetes mellitus tipo 2 (DMT2) en comparación con placebo, cuando se adicionó a la terapia basal actual (Relación de Riesgo [RR] 1.00; intervalo de confianza de 95% [IC]: 0.89, 1.12; P < 0.001 para no inferioridad). (Ver Estudios clínicos). En el estudio SAVOR, la incidencia de eventos de pancreatitis relacionados fue de 0.3% tanto en los pacientes tratados con ONGLYZA como en los pacientes tratados con placebo en la población con intención de tratamiento. La incidencia de reacciones de hipersensibilidad fue de 1.1% tanto en los pacientes tratados con ONGLYZA como en los pacientes tratados con placebo. Hipoglucemia: En el estudio SAVOR, la incidencia total reportada de hipoglucemia (registrada en el diario del paciente) fue de 17.1% en los pacientes tratados con ONGLYZA y 14.8% en los pacientes tratados con placebo. El porcentaje de pacientes con eventos hipoglucémicos graves reportados durante el tratamiento (definido como un evento que requirió la ayuda de otra persona) fue mayor en el grupo de saxagliptina que en el grupo de placebo (2.1% y 1.6%, respectivamente). El riesgo incrementado de hipoglucemia total y grave observado en el grupo tratado con saxagliptina ocurrió principalmente en pacientes tratados con una sulfonilurea al inicio y no en pacientes tratados con insulina o monoterapia de metformina al inicio. El riesgo incrementado de hipoglucemia total y grave se observó principalmente en pacientes con HbA1C < 7% al incio. Reacciones adversas relacionadas con ONGLYZA® en estudios de control glucémico: Existieron 4148 pacientes con diabetes tipo 2 distribuidos aleatoriamente, incluyendo 3021 pacientes tratados con ONGLYZA®, en seis estudios de seguridad y eficacia clínica controlados, con doble ciego, realizados para evaluar los efectos de ONGLYZA® sobre el control glucémico. En un análisis especificado previamente, acumulado de los dos estudios de monoterapia, el estudio de adición a metformina, el estudio de adición a TZD y el estudio de adición a glibenclamida, la incidencia total de los eventos adversos en pacientes tratados con 5 mg de ONGLYZA®, fue similar al de placebo. La discontinuación de la terapia debida a eventos adversos fue más alta en pacientes que recibieron 5 mg de ONGLYZA® en comparación a placebo (3.3% en comparación a 1.8%). Las reacciones adversas reportadas en pacientes tratados con ONGLYZA® se muestran en la Tabla 8. Las reacciones adversas son enlistadas por clase de órganos del sistema y por la frecuencia absoluta. Las frecuencias son definidas como muy común (≥1/10), común (≥1/100, < 1/10), poco común (≥1/1000, < 1/100), raro (≥1/10,000, < 1/1000), o muy raro ( < 1/10,000).

En el análisis combinado de cinco estudios, un grupo de eventos relacionados con hipersensibilidad mostró hasta la semana 24 una incidencia de 1.5 % y 0.4% en los pacientes que recibieron ONGLYZA® 5 mg y placebo, respectivamente. En los pacientes que recibieron ONGLYZA®, ninguno de estos eventos requirió hospitalización o que fuera reportado por los investigadores por poner la vida en peligro. Reacciones Adversas Asociadas con ONGLYZA® y Terapia Concomitante en estudios de Control Glucémico: En el estudio de adición a glibenclamida, fue poco común la incidencia de hipoglucemia confirmada (definida como síntomas de hipoglucemia acompañada de glucosa capilar ≤50 mg/dL) para los grupos de 5 mg de ONGLYZA® (0.8%) y placebo (0.7%). La diferencia no fue estadísticamente significativa. En los dos estudios de monoterapia, el estudio de adición a metformina, y el estudio de adición a tiazolidinediona (TZD), la incidencia de las reacciones adversas de hipoglucemia confirmada en pacientes tratados con 5mg de ONGLYZA® fue similar al placebo. En el estudio de adición a insulina, la incidencia total de hipoglucemia reportada fue 18.4% para ONGLYZA® 5 mg y 19.9% para placebo. La incidencia de hipoglucemia confirmada para ONGLYZA® 5mg fue 5.3% versus 3.3% para placebo. En el estudio de adición de la combinación de metformina más SU, la incidencia total de hipoglucemia reportada fue de 10.1% para ONGLYZA® 5 mg y de 6.3% para placebo. La hipoglucemia confirmada fue reportada en 1.6% de los pacientes tratados con ONGLYZA® y ninguna para pacientes tratados con placebo. En el estudio de adición a TZD, la incidencia de edema periférico fue común y más alta para 5 mg de ONGLYZA® en comparación a placebo (8.1% en comparación a 4.3%). Todas las reacciones adversas reportadas de edema periférico fueron de intensidad leve a moderada y ninguna dio como resultado la discontinuación del fármaco de estudio. En un análisis acumulado de los dos estudios de monoterapia, el estudio de adición a metformina, y el estudio de adición a SU, la incidencia general de las reacciones adversas de edema periférico observado en pacientes tratados con 5 mg de ONGLYZA®fue similar al placebo (1.7% en comparación a 2.4%). En la terapia inicial de combinación de ONGLYZA® 5 mg y metformina, la incidencia de nasofaringitis fue común y más alta para ONGLYZA® más metformina (6.9%) en comparación a 10 mg de saxagliptina (4.2%) y metformina sola (4.0%). La incidencia de la cefalea fue común y más alta para ONGLYZA® 5 mg más metformina (7.5%) en comparación a 10 mg de saxagliptina (6.3%) y metformina sola (5.2%). Experiencias Post-comercialización: Las siguientes reacciones adversas se han reportado con el uso de saxagliptina durante la etapa de postcomercialización: pancreatitis aguda y reacciones de hipersensibilidad, incluyendo anafilaxia, angioedema, exantema y urticaria. Puesto que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no es posible estimar su frecuencia de manera confiable (Ver Contraindicaciones).
Interacciones medicamentosas y de otro género: El metabolismo de saxagliptina está principalmente mediado por el citocromo P450 3A4/5 (CYP3A4/5). En estudios in vitro, saxagliptina y su metabolito principal no inhibieron CYP1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1 o 3A4, ni indujeron CYP1A2, 2B6, 2C9 o 3A4. Por tanto, no se espera que saxagliptina altere la depuración metabólica de los fármacos coadministrados que son metabolizados por estas enzimas. Saxagliptina tampoco es un inhibidor significativo de la P-glucoproteína (P-gp) ni tampoco un inductor de P-gp. La unión con proteínas de saxagliptina y de su metabolito principal, in vitro, en el suero humano, está por debajo de los niveles cuantificables. Por tanto, la unión a proteínas podría no tener una influencia significativa sobre la farmacocinética de saxagliptina, o de otros fármacos. Efecto de otros fármacos sobre saxagliptina: En estudios realizados en sujetos sanos, como se describe más adelante, la farmacocinética de saxagliptina, su metabolito principal, o la exposición a los componentes activos totales de saxagliptina (original + metabolito), no fueron significativamente alterados por metformina, glibenclamida, pioglitazona, digoxina, simvastatina, diltiazem, ketoconazol, rifampicina, omeprazol, hidróxido de aluminio + hidróxido de magnesio + simeticona en combinación, o famotidina. Metformina: La coadministración de una dosis única de saxagliptina (100 mg) y metformina (1000 mg), un sustrato de hOCT-1 y hOCT-2, disminuyó la Cmax de saxagliptina en 21%; no obstante, el AUC permaneció sin cambio. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otro sustrato de hOCT-1 y hOCT-2. Glibenclamida: La coadministración de una dosis única de saxagliptina (10 mg) y glibenclamida (5 mg), un sustrato de CYP2C9, incrementó la Cmax de saxagliptina en 8%; sin embargo, el AUC de saxagliptina permaneció sin cambio. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otros sustratos de CYP2C9. Pioglitazona: La coadministración de varias dosis una vez al día de saxagliptina (10 mg) y pioglitazona (45 mg), un sustrato de CYP2C8 (mayor) y CYP3A4 (menor), no alteró la farmacocinética de saxagliptina. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otros sustratos de CYP2C8. Digoxina: La coadministración de varias dosis una vez al día de saxagliptina (10 mg) y digoxina (0.25 mg), un sustrato de P-gp, no alteró la farmacocinética de saxagliptina. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otros sustratos de P-gp. Simvastatina: La coadministración de varias dosis una vez al día de saxagliptina (10 mg) y simvastatina (40 mg), un sustrato de CYP3A4/5, incrementó la Cmax de saxagliptina en un 21%; sin embargo, el AUC de saxagliptina permaneció sin cambio. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otros sustratos de CYP3A4/5. Diltiazem: La coadministración de una dosis única de saxagliptina (10 mg) y diltiazem (formulación de acción prolongada de 360 mg en fase estable), un inhibidor moderado de CYP3A4/5, incrementó la Cmax para saxagliptina un 63%, y el AUC para los componentes activos totales de saxagliptina en un 21%. Por tanto, no se esperan interacciones significativas de ONGLYZA® con otros inhibidores moderados de CYP3A4/5. Ketoconazol: La coadministración de una dosis única de saxagliptina (100 mg) y ketoconazol (200 mg cada 12 horas en fase estable), un inhibidor potente de CYP3A4/5 y P-gp, incrementó la Cmax para saxagliptina en un 62% y el AUC para los componentes activos totales de saxagliptina en un
Alteraciones en los resultados de pruebas de laboratorio: Entre estudios clínicos, la incidencia de eventos adversos de laboratorio fue similar en pacientes tratados con 5 mg de ONGLYZA® comparada con la de los pacientes tratados con placebo. Fue observada una pequeña disminución en la cuenta absoluta de linfocitos. A partir de un valor basal de la cuenta absoluta de linfocitos de aproximadamente 2200 células/microlitro, se observó una disminución media de aproximadamente 100 células/microlitro comparado a placebo, en un análisis combinado de cinco estudios clínicos controlados con placebo. Las medias de cuentas absolutas de linfocitos permanecieron estables y dentro de los límites normales con una dosificación diaria de hasta 102 semanas de duración. Las disminuciones en la cuenta de linfocitos no se asociaron con reacciones adversas clínicamente relevantes. No se conoce la significancia clínica de esta disminución en la cuenta de linfocitos en comparación al placebo. En el estudio SAVOR, se reportó reducción del recuento de linfocitos en 0.5% de los pacientes tratados con ONGLYZA® y 0.4% en los pacientes tratados con placebo.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis, Mutagénesis, Alteraciones en la Fertilidad: Se realizaron estudios de carcinogenicidad de dos años en ratones y ratas a dosis orales de 50, 250 y 600 mg/kg/día y 25, 75, 150 y 300 mg/kg/día, respectivamente. Saxagliptina no indujo tumores en ratones o en ratas a las dosis más altas evaluadas. Las dosis más altas evaluadas en ratones fueron equivalentes aproximadamente a 900 (machos) y 1210 (hembras) veces la exposición humana a la dosis humana recomendada de 5 mg/día (RHD). En ratas, las exposiciones fueron de aproximadamente 370 (machos) y 2300 (hembras) veces la RHD. Fue probado el potencial mutagénico y clastogénico de saxagliptina a altas concentraciones y exposiciones en una batería de estudios de toxicidad genética que incluyen un ensayo bacteriano de Ames in vitro, un ensayo de citogenética in vitro en linfocitos humanos primarios, un ensayo de micronúcleos orales in vivo en ratas, un estudio de reparación del ADN oral in vivo en ratas y un estudio de citogenética oral in vivo/in vitro en linfocitos de sangre periférica de rata. Saxagliptina no fue mutagénica o clastogénica con base en los resultados combinados de estos estudios. El metabolito principal no fue mutagénico en un ensayo bacteriano de Ames in vitro. En un estudio de fertilidad en rata, se trataron machos con dosis de alimentación por sonda vía oral de 100, 200 y 400 mg/kg/día por dos semanas antes de la cópula, durante la cópula y hasta la terminación programada (aproximadamente cuatro semanas en total) y las hembras fueron tratadas con dosis de alimentación por sonda vía oral de 125, 300, y 750 mg/kg/día por dos semanas antes de la cópula hasta el día 7 de la gestación. No se observaron efectos adversos sobre la fertilidad a 200 mg/kg/día (machos) o 125 mg/kg/día (hembras) dando como resultado exposiciones respectivas (AUC) de aproximadamente 630 (machos) y 805 (hembras) veces la exposición humana a la RHD. A dosis maternalmente tóxicas más altas (300 y 750 mg/kg/día), se observaron reabsorciones fetales incrementadas (aproximadamente 2150 y 6375 veces la RHD). Se observaron efectos adicionales sobre el ciclo estrogénico, fertilidad, ovulación e implantación a 750 mg/kg (aproximadamente 6375 veces la RHD). Teratogenicidad y Deterioro del Desarrollo Fetal: Saxagliptina no fue teratogénica a ninguna dosis evaluada en ratas o conejos. A altas dosis en ratas, saxagliptina provocó un retraso menor y reversible del desarrollo en la dosificación de la pelvis fetal a ≥240 mg/kg/día (≥1560 veces la exposición humana [AUC] a la RHD). La toxicidad materna y los pesos corporales fetales reducidos se observaron a 900 mg/kg/día (8290 veces la RHD). En conejos, los efectos de saxagliptina estuvieron limitados a variaciones esqueléticas menores observadas únicamente a dosis maternalmente tóxicas (200 mg/kg/día, exposiciones 1420 veces la RHD). Saxagliptina administrada a ratas hembra del día de gestación 6 hasta el día de la lactancia 20 dio como resultado pesos corporales disminuidos en las crías de macho y hembra únicamente a dosis maternalmente tóxicas (≥250 mg/kg/día, exposiciones ≥1690 veces la RHD). No fue observada toxicidad funcional o conductual en las crías de ratas tratadas con saxagliptina a cualquier dosis. Toxicología Animal: Saxagliptina produjo cambios en la piel (costras y/o ulceraciones) en las extremidades (cola, dedos, escroto, y/o nariz) e infiltración de células mononucleares en órganos múltiples, microscópica e inflamación en los monos cynomolgus a dosis ≥2 mg/kg/día por 1 a 3 meses (≥7 veces la exposición en humanos a la RHD). En un estudio de tres meses, a 3 mg/kg/día (≥20 veces la exposición a la RHD), fue observada la curación de la piel durante el periodo de dosificación, con recuperación completa de la piel y los cambios microscópicos después de un periodo de recuperación libre de fármaco. Los infiltrados de células mononucleares o la inflamación son considerados como una exacerbación de los cambios de fondo comúnmente observados en monos. El nivel sin efecto para la piel y los cambios microscópicos es 0.3 mg/kg/día (una [hembra] a tres [machos] veces la RHD). Lesiones similares de la piel no han sido observadas en ratones, ratas o perros a exposiciones hasta de 1210, 2300 ó 55 veces la exposición de humanos a la RHD, respectivamente. No han sido observadas correlaciones clínicas a las lesiones en piel en monos, en las pruebas clínicas en humanos con saxagliptina. Saxagliptina produjo heces sanguinolentas/mucoides en perros (a exposiciones ≥19 veces la RHD) con un nivel de cuatro veces sin efecto de la RHD.
Dosis y vía de administración: Dosis Recomendada: ONGLYZA® tabletas no debe ser triturada ni masticada. Monoterapia y Terapia de Combinación Adicionada: La dosis recomendada de ONGLYZA® es 5 mg una vez al día como monoterapia o como terapia de combinación adicionada a metformina o a tiazolidinediona o a sulfonilurea o a insulina (con o sin metformina). ONGLYZA® puede ser tomada con o sin los alimentos. Terapia de Combinación Inicial: Las dosis iniciales recomendadas de ONGLYZA® y metformina, cuando se utilizan como terapia de combinación inicial, son de 5 mg de ONGLYZA® más 500 mg de metformina una vez al día. Los pacientes con control glucémico inadecuado con estas dosis iniciales deberán incrementar las dosis de metformina basados estrictamente en la recomendación del clínico. Insuficiencia Renal: La evaluación de la función renal se recomienda antes del inicio con ONGLYZA® y posteriormente de una manera periódica. (Ver Precauciones Generales y Farmacocinética y Farmacodinamia). Insuficiencia Renal Leve: No se requiere ajuste de dosis para pacientes con insuficiencia renal leve (TFG estimada 60-89 mL/min/1.73 m2, por dieta modificada en Enfermedad Renal ecuación MDRD TFG estimada. Insuficiencia Renal Moderada: No se requiere ajuste de dosis para pacientes con TFG ≥45mL/min/1.73m2: Para pacientes con insuficiencia renal moderada con < 45mL/min/1.73m2, la dosis es 2.5mg una vez al día. Insuficiencia renal severa: Para pacientes con insuficiencia renal severa (TFG estimada < 30mL/min/1.73m2) o con Insuficiencia Renal Terminal (ESRD) que requieren hemodiálisis, la dosis es 2.5mg una vez al día. ONGLYZA® debe ser administrada después de la hemodiálisis. ONGLYZA® no ha sido estudiada en pacientes que reciben diálisis peritoneal. Insuficiencia hepática: No es necesario ningún ajuste de la dosis para ONGLYZA® en pacientes con insuficiencia hepática leve, moderada o severa. Pediátrico y adolescente: La seguridad y la efectividad de ONGLYZA® en pacientes pediátricos y adolescentes menores de 18 años no han sido establecidas, por lo que no se recomienda. Geriátrica: No se requiere ningún ajuste para ONGLYZA® con base únicamente en la edad. Debido a que es más probable que los pacientes de edad avanzada tengan función renal disminuida, se debe tener cuidado en la selección de la dosis en estos sujetos, con base en la función renal (Ver Precauciones Generales).
Manifestaciones y manejo de la sobredosificación o ingesta accidental: ONGLYZA®oralmente administrada, una vez al día, ha mostrado ser segura y bien tolerada, sin efecto clínicamente significativo sobre el intervalo de QTc o el ritmo cardiaco a dosis de hasta 400 mg diariamente por dos semanas (80 veces la dosis recomendada humana de 5 mg/día (RHD). En el caso de una sobredosis, el tratamiento de apoyo apropiado debe ser iniciado según esté dictado por el estado clínico del paciente. Saxagliptina y su metabolito principal son eliminados mediante hemodiálisis (23% de la dosis en cuatro horas).
Presentación(es): Caja con 28 tabletas de 2.5 m. Caja con 14 y 28 tabletas de 5 mg.
Recomendaciones sobre almacenamiento: Consérvese no más de 30°C.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. No se use durante el embarazo, lactancia y menores de 18 años. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y al correo patientsafety.mexico@astrazeneca.com.
Nombre y domicilio del laboratorio: AstraZeneca. S.A. de C.V. Super Av. Lomas Verdes No. 67, Fracc. Lomas Verdes, C.P. 53120, Naucalpan de Juárez, México, México.
Número de registro del medicamento: 221M2009 SSA IV.


 Cambiar de país
Cambiar de país