6-UR
SANFER
Denominación genérica: Levetiracetam.
Forma farmacéutica y formulación: Tableta. Cada Tableta contiene: Levetiracetam 500mg y 1000mg Excipiente c.b.p 1 Tableta.
Indicaciones terapéuticas: Levetiracetam está indicado como monoterapia, en el tratamiento de las crisis convulsivas parciales, con o sin generalización secundaria; en pacientes mayores de 16 años con diagnóstico reciente de epilepsia. Levetiracetam está indicado como terapia concomitante: En el tratamiento de la crisis de inicio parcial con o sin generalización secundaria en adultos y niños mayores de 4 años con eplilepsia. En el tratamiento de la crisis nioclónicas en adultos y adolescentes mayores de 12 años con Epilepsia mioclónica Juvenil. En el tratamiento de las crisis tónico-clónicas generalizadas primarias en adultos y adolescentes mayores de 12 años con epilepsia idiopática generalizada.
Farmacocinética y farmacodinamia: Farmacocinética: Levetiracetam es un compuesto altamente soluble y permeable. Su perfil farmacocinético es lineal e independiente del tiempo con baja variabilidad interindividual. No hay modificación en la depuración después de varias dosis administradas. No hay evidencia de variabilidad respecto al género, raza o ritmo circadiano. El perfil farmacocinético es comparable en voluntarios sanos y en pacientes con epilepsia. Dada su absorción completa y lineal, pueden predecirse los niveles plasmáticos en mg/Kg, a partir de una dosis administrada por vía oral. Por lo que no es necesario monitorizar los niveles plasmáticos de Levetiracetam. Se ha demostrado que existe una correlación significativa entre la concentración plasmática y en saliva de Levetiracetam tanto en adultos como en niños (la relación saliva/plasma van de 1 a 1.7 en comprimidos orales y después de 4 horas de la administración de la solución oral). Absorción: Levetiracetam se absorbe rápidamente después de su administración oral. La biodisponibilidad absoluta oral es cercana al 100%. La concentración máxima a nivel plasmático (Cmax) se alcanza 1.3 horas luego de su administración. El pico de concentración (Cmax) es de 31 a 43 mg/ml seguido de una dosis única de 1000mg, o 1000mg repartido dos veces al día. El estado estacionario se alcanza dos días después de su administración repetida de 1000 mg dos veces al día respectivamente. La tasa de absorción es independiente de la dosis y no se altera por la ingesta de alimentos. Distribución: No hay estudios de distribución en tejidos humanos. Ni Levetiracetam ni su metabolito primario se unen significativamente a las proteínas plasmáticas ( < 10%). El volumen de distribución del Levetiracetam es aproximadamente 0.5 a 0.7 l/kg, un valor cercano al volumen total de agua corporal. Metabolismo: Levetiracetam no es metabolizado extensamente por los humanos. Su vía metabólica principal (24% de la dosis) es mediante la hidrólisis enzimática del grupo acetamida. La producción del metabolito primario, ucb L057, no es mediada por las isoformas del sistema citocromo P450 hepático. La hidrólisis del grupo acetamida es cuantificable en varios tejidos del cuerpo, incluido las células sanguíneas. El metabolito ucb L057 es farmacológimanete inactivo. Otros dos metabolitos menores también fueron identificados. Uno se obtuvo por hidroxilación del anillo de pirrolidona (1.6% de la dosis), y el otro mediante la apertura del anillo de pirrolidona (0.9% de la dosis). Otros componentes sin identificar corresponden solo al 0.6% de la dosis. No se observó inter conversión de ningún enantiómero del Levetiracetam o su metabolito primario en la experiencia in-vivo. Los estudios in vitro han demostrado que Levetiracetam y su metabolito principal no inhiben la actividad de las isoformas hepáticas principales del citocromo P450 (CYP3A4, CYP2A6, CYP2C9,CYP2C19, CYP2D6, CYP2E1 Y CYP1A2), de la enzima glucuronil transferasa (UGT1A1 Y UGT1A6) ni la actividad de la epóxido hidrolasa. Además, Levetiracetam no afecta la glucuronidación in vitro del ácido valproico. En cultivos de hepatocitos humanos, Levetiracetam tiene un efecto mínimo en la conjugación del etinilestradiol o del CYP1A2, SULT1E1 O UGT1A1. Levetiracetam provocó una leve inducción del CYP2B6 y del CYP3A4 a concentraciones elevadas (680 ug/ml), sin embargo a concentraciones aproximadas a Cmax, que se alcanza después de aplicaciones repetidas de 1500 mg/2veces/día, los efectos no fueron considerados biológicamente relevantes. Debido a lo anterior es muy poco probable que Levetiracetam interaccione con otras sustancias, o viceversa. Eliminación: La vida media plasmática en adultos fue de 7±1 hora, y no varía con la dosis, vía de administración o administración repetida. El promedio de la depuración corporal total es de 0.96 mL/min./kg. La ruta principal de excreción es por vía urinaria, en un 95% de la dosis y aproximadamente un 0.3% de la dosis es excreción vía fecal. La excreción urinaria acumulada del Levetiracetam ocurre en un 66% durante las primeras 48 horas y de su metabolito primario en un 24% por lo que aproximadamente un 93% de las dosis se elimina en las primeras 48 horas. La depuración renal de Levetiracetam y del ucb LO57 es de 0.6 y de 4.2 mL/min/Kg respectivamente, lo que indica que Levetiracetam es excretado mediante filtración glomerular con la subsecuente reabsorción tubular y que el metabolito primario también se excreta por secreción tubular activa, además de la filtración glomerular. La eliminación del Levetiracetam se correlaciona con la depuración de creatinina. Poblaciones especiales de pacientes: Niños (4 a 12 años): después de una administración única (20mg/kg) a niños con diagnóstico de epilepsia, la vida media del Levetiracetam fue de 6 horas: La depuración corporal aparente aajustado al peso fue de 1.4 mL/min/Kg depués de la administración repetida de dosis orales (20 a 60 mg/Kg/día) a niños epilépticos (de 4 a 12 años), Levetiracetam fue absorbido rápidamente. El pico máximo de concentración plasmática se observó de 0.5 a 1 hora después de su administración. Se observaron incrementos lineales y proporcionales a la dosis para el pico de concentracion plasmática y el área bajo la curva. La vida media de eliminación es de aproximadamente 5 horas. La depuración corporal aparente fue de 1.1 mL/min/Kg. Ancianos: En los ancianos, la vida media incrementa en aproximadamente un 40% (10 a 11 horas). Esto se relaciona con la disminución de la función renal en esta población. Insuficiencia renal: La depuración renal aparente de ambos, del Levetiracetam y su metabolito primario se correlaciona con la depuración de creatinina. Por lo que se recomienda ajustar la dosis diaria de mantenimiento de Levetiracetam basado en la depuración de creatinina en pacientes con insuficiencia renal moderada y severa. En pacientes con anuria en fase terminal de falla renal, la vida media del fármaco fue aproximadamente desde 25 a 3 horas en periodos interdiálitico e intradiálitico, respectivamente. La remoción fraccional de Levetiracetam durante una sesión de diálisis de 4 horas fue de 51%. Insuficiencia hepática: En sujetos con insuficiencia hepática leve o moderada, no hubo una modificación relevante de la depuración de Levetiracetam. En la mayoría de los sujetos con insuficiencia hepática severa, la depuración renal disminuida concomitante, explica la mayor parte de esta disminución. Farmacodinamia:Mecanismo de acción: El principio activo, Levetiracetam, es un derivado de la pirrolidona (S-enatiómero de a-etil-2-oxo-1-pirrolidina acetamida), no relacionado quimicamente con las sustancias activas de los antiepilépticos existentes en la actualidad. El mecanismo de acción del Levetiracetam aun no ha sido totalmente dilucidado, pero parece ser diferente a los mecanismos de los otros medicamentos antiepilépticos existentes en la actualidad. Los estudios in-vitro muestran que Levetiracetam afecta los niveles de Ca2+ intraneuronal por inhibición parcial de las corrientes de Ca2+ de tipo N y por reducción de la liberación de Ca2+ de los almacenes intraneuronales. Adicionalmente, revierten parcialmente las reducciones de las corrientes generadas por GAB, y glicina inducidas por zinc y b-carbonilos. Un estudio in-vitro mostró que Levetiracetam se une a un punto específico del tejido cerebral de roedores. Este sitio específico es la proteína 2A de las vesículas sinápticas, la cual se cree que está involucrada en la fusión de las vesículas y la liberación de neurotransmisores. Levetiracetam y los análogos relacionados muestran un rango de afinidad para la unión de la proteína 2A de las vesículas sinápticas que se correlaciona con la potencia de su protección anticonvulsiva en el modelo audiogénico de epilepsia del ratón. Este hallazgo sugiere que la interacción entre el Levetiracetam y la proteína 2A de la vesícula sináptica parece contribuir al mecanismo de acción antiepiléptico del fármaco. Efectos farmacodinámicos: Levetiracetam induce protección sobre la crisis convulsivas en un amplio rango de modelos animales de crisis generalizadas primeras y paarciales sin tener un efecto pro convulsivo. El metabolito principal es inactivo. En humanos, la actividad en epilepsia parcial y generalizada (descargas epileptiformes/respuesta foto paroxóstocas) han confirmado el amplio espectro de su perfil farmacológico preclínico.
Contraindicaciones: Hipersensibilidad a Levetiracetam, o a otros derivados de los pirrolidonas o a cualquiera de los componentes de la fórmula. No debe utilizarse en el embarazo y la lactancia, menores de 16 años como monoterapia.
Precauciones generales: De acuerdo con las prácticas clínicas actuales, si Levetiracetam debe descontinuarse, se recomienda que el retiro se realice gradualmente (por ejemplo, en adultos y adolescentes de 50 Kg de peso o más: reducciones de 500 mg 2 veces al día cada dos a cuatro semanas; en los niños y adolescentes que pesen menos de 50 Kg: la disminución de la dosis no debe exceder los 10 mg/Kg 2 veces al día cada dos semanas). La administración de Levetiracetam a pacientes con insuficiencia renal puede requerir ajuste en sus dosis. En pacientes con disfunción hepática severa, se recomienda un estudio de la función renal antes de seleccionar la dosis. Se han reportado suicidios, intentos de suicidio y/o ideas suicidas en pacientes tratados con Levetiracetam, por lo que los pacientes deben ser informados de que en caso de presentar cualquier síntoma de depresión y/o ideas suicidas, avisar inmediatamente a su médico tratante. Debido a las diferencias individuales en la sensibilidad, algunos pacientes pueden presentar al inicio dei tratamiento o con el aumento progresivo de la dosis, somnolencia u otros síntomas relacionados con el sistema nervioso central. Por lo que se recomienda precaución a los pacientes que realicen actividades que les pongan en riesgo. Efectos sobre capacidad de conducir y utilizar maquinarias: No se ha realizado ningún estudio sobre la capacidad de conducir u utilizar maquinas. Debido a la posible diversa sensibilidad individual, algunos pacientes pueden experimentar somnolencia u otros síntomas relacionados al sistema nervioso central, al inicio del tratamiento o después de un aumento de la dosis. Por lo tanto, se recomienda tener precaución en estos pacientes al realizar tareas calificadas.
Restricciones de uso durante el embarazo y lactancia: No hay datos disponibles del uso de Levetiracetam en mujeres embarazadas. Estudios en animales han mostrado toxicidad reproductiva. Se desconoce el riesgo potencial en humanos. No debe administrarse en el embarazo, a menos que sea claramente necesario. Como con el uso de otros fármacos antiepilépticos, los cambios fisiológicos que ocurren durante el embarazo pudieran afectar la concentración plasmática de Levetiracetam. Sin embargo, no se han encontrado reportes que demuestren la disminución de la concentración plasmática de Levetiracetam durante el embarazo. El descontinuar el tratamiento antiepiléptico puede resultar en una exacerbación de la enfermedad lo que puede resultar perjudicial para la madre y el feto. Lactancia: Levetiracetam es excretado en la leche materna humana. Por lo tanto, no se recomienda la lactancia durante su uso.
Reacciones secundarias y adversas: En estudios clínicos controlados con una participación de 1,023 pacientes con epilepsia. Los datos de seguridad reunidos de estos estudios en pacientes adultos demostraron que 46.4% y 42.2% de los pacientes experimentaron efectos indeseables en los grupos con Levetiracetam y placebo respectivamente y que 2.4% y 2% de los pacientes sufrieron efectos adversos serios en los grupos con Levetiracetam y placebo respectivamente. Los eventos adversos reportados más comunmente fueron somnolencia, astenia y mareos, en un estudio controlado con pacientes pediátricos (de 4 a 16 años de edad) mostró que 55.4% de los pacientes en el grupo tratado con Levetiracetam y 40.2% de los pacientes en el grupo placebo experimentaron efectos indeseables. Los eventos adversos serios fueron experimentados por 0.0% de los pacientes en el grupo tratado con Levetiracetam y 1.0% de los pacientes del grupo placebo. Los efectos indeseables más comúnmente reportados en la población pedriática fueron somnolencia, hostilidad, nerviosismo, inestabilidad emocional, agitación, anorexia, astenia y cefalea. Los resultado de seguridad de Levetiracetam son generalmente similares entre los grupos de edad (pacientes adultos y pediátricos) y entre las indicaciones para epilepsia aprobadas. No hubo evidencia de relaciones dosis respuesta en la severidad de eventos adversos relacionados con el sistema nervioso central. En un estudio realizado en niños y adultos (4 a 65 años) con epilepsia generalizada idiopática con convulsiones tónico clónicas generalizadas primarias se encontró, que el 39.2% de los pacientes en el grupo de Levetiracetam y el 29.8% de los pacientes en el grupo con placebo, presentaron efectos indeseables que se consideraron relacionados con el tratamiento. El más comunmente reportado fue fatiga. La incidencia de efectos indeseables (eventos adversos considerados posiblemente a Levetiracetam por el investigador) de estudios clínicos controlados fueron: Efectos indeseables muy comunes ( > 10%) Trastornos generales y condición del sitio de administración: astenia. Trastornos del sistema nervioso: somnolencia/fatiga. Efectos indeseables comunes ( > 1%, < 10%): Trastornos del sistema sanguíneo: Trombocitopenia Trastornos gastrointestinales: dolor abdominal, diarrea, dispepsia, náusea, vómito. Trastornos del sistema nervioso: amnesia, ataxia, convulsiones, cefalea, hiperquinesia, temblor, trastornos del equilibrio, problemas de atención, pérdida de la memoria. Trastornos psiquiátricos: agresión, agitación, depresión, debilidad emocional/cambio de humor, hostilidad, insomnio, irritabilidad, nerviosismo, trastornos de la personalidad, pensamientos anormales. Trastornos del metabolismo y la nutrición: anorexia, incremento de peso (el riesgo de anorexia es mayor cuando se administra Levetiracetam en combinación con Topiramato). Trastornos del oído y del laberinto: vértigo. Trastornos de la visión: diplopía y visión borrosa. Trastornos del sistema músculo-esqueletico y del tejido conectivo: mialgias. Trastornos respiratorios torácicos y mediastinales: exacerbación de tos. Trastornos de la piel y tejido subcutáneo: eccema, prurito, erupción. Experiencia post comercialización: las alteraciones psiquiátricas o del sistema nervioso son los datos más frecuentemente reportados. Además de los eventos adversos reportados durante el desarrollo de estudios clínicos, los siguientes eventos adversos han sido reportados en la experiencia post comercialización. Los datos son insuficientes para soportar un estimado de su incidencia en la población a ser tratada. Trastornos de la sangre y dei sistema linfático: leucopenia, neutropenia, pancitopenia (con la supresión de medula identificada en algunos casos). Trastornos psiquiátricos: comportamiento anormal, cólera, ansiedad, confusión,
alucinación, trastornos psicóticos, suicidios, intentos de suicidio y/o de ideas suicidas. Trastornos del sistema nervioso: parestesia. Trastornos gastrointestinales: pancreatitis. Trastornos de la piel y tejido subcutáneo: alopecia en varios casos; se observó recuperación cuando Levetiracetam fue suspendido.
Interacciones medicamentosas y de otro género: Datos de estudios clínicos indican que Levetiracetam no influye en las concentraciones séricas de otros fármacos antiepilépticos conocidos (como Fenítoína, Carbamazepina, ácido Valproico, Fenobarbital, Lamotrigina, Gabapentina y Primidona) y que estos medicamentos antiepilépticos no influyen en la famnacocinética de Levetiracetam. La depuración de Levetiracetam fue 22% mayor en niños que toman antiepilépticos inductores de enzimas, al comparase con aquellos tratados con antiepilépticos no inductores enzimáticos, por lo que no se recomienda el ajuste de dosis. Ei Levetiracetam no afecta las concentraciones plasmáticas de Carbamacepina, Valproato, Topiramato y Lamotrigina. Se ha comprobado que con Probenecid (500 mg 4 veces al día), bloqueador de la secreción tubular renal, inhibe la depuración renal del metabolito primario pero no del Levetiracetam. Sin embargo, la concentración de este metabofito permanece baja. Es esperable que otros productos que son excretados por secreción tubular activa también pudieran disminuir la depuración renal del metabolito. El efecto de Levetiracetam sobre el Probenecid no ha sido estudiado. No se conoce el efecto de Levetiracetam en otros productos secretados activamente, como por ejemplo AINES, Sulfonamidas y Metotrexato. Dosis diarias de 1000 mg de Levetiracetam no influyen en la farmacocinética de anticonceptivos orales, (Etinil-Estradiol y Levonorgestrel); los parámetros endocrinos (hormona Luteinizante y Progesterona) no fueron modificados. Dosis diarias de 2000 mg de Levetiracetam no influencian la farmacocinética de la Digoxina y de la Warfarina; el tiempo de protrombina no fue modificado. La coadministración con Digoxina, anticonceptivos orales y Warfarina no influencian la farmacocinética de Levetiracetam. No hay datos disponibles sobre la interacción de Levetiracetam con alcohol.
Alteraciones de pruebas de laboratorio: No se han reportado a la fecha, aunque en estudios clínicos se han observado anormalidades en las pruebas de funcionamiento hepático.
Precaución en relación a efectos de carcinogénesis, mutagénesis y teratogénesis y sobre la fertilidad: Carcinogénesis: Los datos preclínicos no revelan ningún peligro especial para los humanos basados en estudios convencionales de seguridad, farmacología, genotoxicidad y carcinogenicidad. Los efectos adversos no se observaron en estudios clínicos, pero si se observaron en estudios realizados en rata y en menor grado en ratón, cuando se sometieron a niveles similares a los niveles de exposición en humanos, y con posible relevancia para uso clínico fueron, cambios en hígado, que indicaban una respuesta adaptativa incrementada, así como hipertrofia centrilobular, infiltración grasa e incremento de las enzimas del hígado en plasma. Las ratas fueron dosificadas con Levetiracetam en la dieta durante 104 semanas a dosis de 50, 300 y 1,800 mg/Kg/día. La dosis más alta corresponde a 6 veces la dosis máxima en humanos recomendada (MRHD) de 3,000 mg sobre una base de mg/m2 y esta también proporciona una exposición sistémica (AUC) aproximadamente 6 veces lo alcanzado en humanos que recibían MRHD. No hay evidencia de carcinogenicidad. Han sido realizados dos estudios en ratones. En un estudio, los ratones recibieron Levetiracetam en la dieta durante 80 semanas en dosis de 60, 240 y 960 mg/Kg/día (la dosis alta es equivalente a 2 veces el MRHD sobre mg/m2 o base de exposición). En un segundo estudio, los ratones recibieron Levetiracetam por vía oral con sonda durante 2 años a niveles de dosis de 1,000, 2,000 y 4,000 mg/Kg/día. Debido a la baja sobrevivencia en la dosis más alta, 4,000 mg/Kg/día en este estudio, la dosis alta fue reducida a 3,000 mg/Kg/día (equivalente a 12 veces el MRHD). Ni unos ni otros estudios demostraron evidencia de carcinogenicidad. En estudios de toxicidad reproductiva en la rata, el Levetiracetam indujo toxicidad durante el desarrollo (incremento en las variaciones o anomalías menores del esqueleto, crecimiento retardado, aumento en la mortalidad fetal) a niveles de exposición similar o mayores a los de exposición humana. En el conejo, los efectos fetales (muerte embrionaria, incremento de anormalidades esqueléticas y de malformaciones) se observaron en los casos de toxicidad materna. La exposición sistemática a nivel de no efecto observado en el conejo fue aproximadamente 4 a 5 veces e! nivel de exposición en humano. Los estudios en animales neonatos y jóvenes (ratas y perros) demostraron que no se observaron efectos adversos en ninguno de los puntos clave de desarrollo o madurez a dosis mayores a los 1,800 mg/Kg/día, correspondiente a 30 veces el máximo de la dosis humana recomendada.
Dosis y vía de administración: Los comprimidos se administran por vía oral, con una cantidad suficiente de líquido y pueden administrarse con o sin alimentos. La posología diaria se divide en dosis iguales repartidas en dos tomas al día. Monoterapia: La dosis inicial recomendada son 250 mg dos veces al día, la cual debe incrementarse hasta la dosis terapéutica inicial de 500 mg 2 veces al día tras dos semanas de tratamiento. La dosis puede ser incrementada por 250 mg dos veces al día cada dos semanas dependiendo de la respuesta clínica observada. La dosis máxima recomendada es de 1500 mg dos veces al día. Terapia concomitante: Adultos (mayores a 18 años) y adolescentes (de 12 a 17 años)con un peso de 50 Kg o superior: La dosis terapéutica inicial es de 500 mg 2 veces ai día. Esta dosis puede ser iniciada desde el primer día de tratamiento. Dependiendo de la respuesta clínica y de la tolerancia, la dosis diaria se puede incrementar hasta 1500 mg 2 veces al día. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg 2 veces al día cada dos a cuatro semanas. Ancianos: Se recomienda ajustar la dosis en pacientes ancianos con función renal comprometida (ver pacientes con insuficiencia renal) Niños: de 4 a 11 años de edad y adolescentes (de 12 a 17 años) con un peso inferior a 50 Kg: La dosis terapéutica inicial es de 10 mg/Kg 2 veces al día. En función de la respuesta clínica y de la tolerabilidad, se puede aumentar la dosis hasta los 30 mg/Kg 2 veces al día. Los cambios de dosis no deberían exceder de los incrementos o reducciones de 10 mg/Kg 2 veces al día cada dos semanas. Se debe utilizar la menor dosis eficaz. La dosificación en niños con un peso de 50 kg o superior es la misma que en adultos. Levetiracetam no está recomendado para uso en niños menores de 4 años debido a la escasez de datos sobre seguridad y eficacia.
Pacientes con Insuficiencia renal: La dosis diaria de Levetiracetam debe individualizarse dependiendo de la función rena!. Para pacientes adultos, referirse a la siguiente tabla y ajustar la dosis indicada. Para usar esta tabla de dosificación, es necesaria una estimación de la depuración de creatinina en ml/min (CLcr).EI CLcr en ml/min puede ser estimado a partir de la determinación de la creatínina sérica (mg/dl), para adultos y adolescentes que pesan 50 Kg o más, a partir de la siguiente fórmula: CLcr (ml/min)= [(140-edad (años)) x peso (Kg)]/[ 72 x creatinina sérica (mg/dl)] (x 0.85 para mujeres). Entonces se ajusta el CLcr para el área de la superficie corporal (ASC) como sigue: CLcr (ml/min/1.73 m2)= [Cl cr (mi/min)/ASC del sujeto (m2)]x1.73.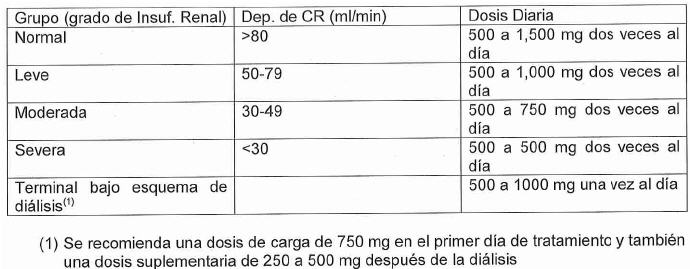
Insuficiencia hepática: No es necesario un ajuste de dosis en pacientes con insuficiencia hepática leve a moderada. En pacientes con insuficiencia hepática grave, la depuración de creatinina puede subestimar el grado de insuficiencia renal, por lo tanto, se recomienda una reducción del 50% de la dosis de mantenimiento diario cuando la depuración de creatinina sea menor de 70 ml/min.
Manifestación y manejo de la de la sobredosificación o ingesta accidental: Síntomas: Se han observado casos de somnolencia, agitación, agresión, depresión del nivel de conciencia, depresión respiratoria y coma por la sobredosis de Levetiracetam.Tratamiento de la sobredosificación: En ia sobredosis aguda puede vaciarse el contenido del estómago por lavado gástrico o por inducción de la émesis. No hay antídoto específico para el Levetiracetam. El tratamiento será sintomático y puede incluir hemodiálisis. La eficacia de la eliminación por diálisis es de 60% para Levetiracetam y de 74% para el metabolito primario.
Presentación(es): Tabletas: Caja con 30 o 60 tabletas de 500 mg. Caja con 30 tabletas de 1000 mg.
Recomendaciones sobre almacenamiento: Consérvese a no más de 25°C.
Leyendas de protección: Su venta requiere receta médica. Prohibida la venta fraccionada del producto. No se use en el embarazo, lactancia, ni menores de 8 años o personas que no puedan deglutir la tableta. No se deje al alcance de los niños. Este medicamento puede producir somnolencia y afectar el estado de alerta, por lo que no deberá conducir vehículos automotores ni maquinaria pesada durante su uso. Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y ucfarmacovigilancia@sanfer.com.mx Literatura exclusiva para el médico.
Nombre y domicilio del laboratorio: Hecho en India por: Intas Pharmaceuticals Limited Sarkhej-bavla Plot 457/458, Matoda, Ahmedabad, Gujarat, 382210, India. Para: Grimann, S.A. de C.V. Circuito Nemesio Diez Riega No. 11, Parque Industrial El Cerrillo II, C.P. 52000, Lerma, México, México. Distribuido por: Laboratorios Sanfer, S.A. De C.V. Hormona No.2 - A, San Andrés Atoto, C.P. 535000, Naucalpan de Juárez, México, México.
Número de Registro del Medicamento: 133M2015 SSA IV