AMIKIN®
BRISTOL M.S.
Denominación genérica: Amikacina
Forma farmacéutica y formulación: Solución inyectable. Cada ampolleta o frasco ámpula contiene: Amikacina 100, 250, 500 o 1,000 mg. Excipiente cbp 2, 2, 2 y 4 ml.
Descripción: Sulfato de amikacina es un antibiótico aminoglucósido semisintético derivado de la kanamicina. AMIKIN® se suministra como una solución incolora, clara y estéril.
Indicaciones terapéuticas: AMIKIN® está indicado en el tratamiento a corto plazo de infecciones graves debidas a cepas susceptibles de bacterias gramnegativas, incluidas especies de Pseudomonas, Escherichia coli, especies de Proteus indol positivias e indol negativas, especies de Providencia, especies de Klebsiella-Enterobacter-Serratia y especies de Acinetobacter (Mima-Herellea). Los estudios clínicos han mostrado que AMIKIN® ha sido efectivo en bacteremia y septicemia (incluida la sepsis neonatal); en infecciones graves del aparato respiratorio, huesos y articulaciones, sistema nervioso central (incluida la meningitis) y piel y tejidos blandos; infecciones intrabdominales (incluida la peritonitis); y en quemaduras e infecciones postoperatorias (incluida la cirugía postvascular). Los estudios clínicos han mostrado que AMIKIN® también es efectivo en infecciones graves, complicadas y recurrentes de las vías urinarias causadas por estos organismos. Los aminoglucósidos, incluido AMIKIN®, no están indicados en episodios iniciales no complicados de infecciones de las vías urinarias a menos que los organismos causales no sean susceptibles a los antibióticos que tiene menos toxicidad potencial. Cuando se indica amikacina en el tratamiento de infecciones no complicadas de las vías urinarias, se puede prescribir una dosificación reducida. (Ver Dosis y vía de administración, Dosis recomendada). Se deben llevar a cabo estudios bacteriológicos para identificar los organismos causantes y su susceptibilidad a la amikacina. AMIKIN® puede considerarse como terapia inicial ante la sospecha de infecciones por gramnegativas y se puede instituir la terapia antes de obtener los resultados de las pruebas de susceptibilidad. Los ensayos clínicos demuestran que AMIKIN® es efectivo en infecciones causadas por cepas resistentes a la gentamicina o tobramicina de organismos gramnegativos, en particular Proteus rettgeri, Providencia stuartii, Serratia marcescens y Pseudomonas aeruginosa. La decisión de continuar la terapia con el fármaco deberá basarse en los resultados de las pruebas de susceptibilidad, la gravedad de la infección, la respuesta del paciente y las consideraciones adicionales importantes incluidas en Precauciones generales, advertencias y precauciones para el uso. AMIKIN® también ha sido efectivo en infecciones por estafilococos y pueden considerarse como terapia inicial en ciertas circunstancias en el tratamiento de enfermedades por estafilococos conocidas o presuntas. Los ejemplos incluyen infecciones graves en las que el organismo causante pudiera ser ya sea una bacteria gramnegativa o un estafilococo, infecciones causadas por cepas susceptibles de estafilococos en pacientes alérgicos a otros antibióticos y en infecciones con mezcla de estafilococos/bacterias gramnegativas. En algunas infecciones graves como sepsis neonatal, la terapia concomitante con un fármaco tipo penicilina podría estar indicado debido a la posibilidad de infecciones causadas por organismos grampositivos como los estreptococos.
Farmacocinética y farmacodinamia: Mecanismo de acción: Microbiología: Gramnegativas: La amikacina es activa in vitro contra especies de Pseudomonas, Escherichia coli, especies de Proteus (indol positivas e indol negativas), especies de Providencia, especies de Klebsiella-Enterobacter-Serratia, especies de Acinetobacter (anteriormente Mima-Herellea) y Citrobacter freundii. Cuando se encuentra que las cepas de los anteriores organismos son resistentes a otros aminoglucósidos, incluidas gentamicina, tobramicina y kanamicina, muchas son susceptibles a la amikacina in vitro. La amikacina resiste la degradación de la mayoría de las enzimas que inactivan al aminoglucósido que se sabe que afectan a la gentamicina, tobramicina y kanamicina. Los estudios in vitro han mostrado que AMIKIN®, en combinación con un antibiótico betalactámico, actúa de manera sinérgica contra muchos de los organismos gramnegativos con importancia clínica. Se presenta la supresión persistente del crecimiento bacteriano de muchos organismos gramnegativos después de la exposición in vitro (efecto post-antibiótico) a AMIKIN®. Grampositivas: La amikacina es activa in vitro contra especies de estafilococos que producen penicilinasa y los que no producen penicilinasa incluidas las cepas resistentes a meticilina. El Staphylococcus aureus resistente a meticilina (MRSA) pudiera no ser totalmente sensible a la amikacina. Los aminoglucósidos en general han mostrado tener una baja actividad contra otros organismos grampositivos, por ejemplo, Streptococcus pyogenes, enterococos y Streptococcus pneumoniae. Pruebas de susceptibilidad de disco: Los métodos cuantitativos que requieren la medición de diámetros de zona proporcionan estimados precisos de susceptibilidad al antibiótico. Uno de estos procedimientos ha sido recomendado para su uso con discos para probar la susceptibilidad a la amikacina. La interpretación involucra la correlación de los diámetros obtenidos en la prueba de disco con valores MIC para la amikacina. Cuando se prueba el organismo causante mediante el método de susceptibilidad en disco de Kirby-Bauer, un disco de 30 mcg de amikacina debe dar una zona de 17 mm o mayor para indicar susceptibilidad. Los tamaños de zona de 14 mm o menores indican resistencia. Los tamaños de zona de 15 a 16 mm indican susceptibilidad intermedia. Con este procedimiento, un informe del laboratorio de "susceptible" indica que el organismo infeccioso es probable que responda a la terapia. Un informe de "resistente" indica que el organismo infeccioso no es probable que responda a la terapia. Un informe de "susceptibilidad intermedia" sugiere que el organismo será susceptible si la infección estuviera confinada a tejidos y líquidos (por ejemplo, orina) en los que se lograran altas concentraciones del antibiótico. Farmacocinética: General: En sujetos adultos normales, la vida media en suero promedio es ligeramente superior a 2 horas con un volumen de distribución aparente total medio de 24 litros, aproximadamente 28 % del peso corporal. La unión a proteínas séricas varía de 0 a 11 %. La velocidad de depuración media en suero es de aproximadamente 100 ml/min y la velocidad de depuración renal es de 94 ml/min en sujetos con función renal normal. La amikacina se excreta principalmente por medio de filtración glomerular. Los pacientes con insuficiencia renal o filtración glomerular disminuida excretan el fármaco de forma mucho más lenta prolongando la vida media en suero. Por lo tanto, se debe supervisar la función renal de manera cuidadosa y por consiguiente se debe ajustar la dosificación. (Ver Dosis y vía de administración, insuficiencia renal). Después de la administración en las dosis recomendadas, se encuentran concentraciones terapéuticas en hueso, corazón, vesícula biliar y tejido pulmonar además de concentraciones significativas en orina, bilis, esputo, secreciones bronquiales y en líquidos intersticial, pleural y sinovial. Los datos de los ensayos con dosis diarias múltiples muestran que las concentraciones del líquido cefalorraquídeo en lactantes normales son de aproximadamente 10 % al 20 % de las concentraciones en suero y pueden alcanzar el 50 % cuando se inflaman las meninges. AMIKIN® ha demostrado que cruza la barrera placentaria y brinda concentraciones importantes en el líquido amniótico. La concentración sérica fetal máxima es de aproximadamente 16 % de la concentración sérica materna máxima y los valores de vida media sérica materna y fetal son de aproximadamente 2 y 3.7 horas, respectivamente. Administración intramuscular: AMIKIN® se absorbe rápidamente y es bien tolerado localmente después de la administración intramuscular. En voluntarios adultos normales, se obtienen concentraciones séricas máximas promedio de aproximadamente 12, 16 y 21 mcg/ml 1 hora después de la administración intramuscular de dosis únicas de 250 mg (3.7 mg/kg), 375 mg (5 mg/kg) y 500 mg (7.5 mg/kg), respectivamente. A las 10 horas, las concentraciones séricas son de aproximadamente 0.3 mcg/ml, 1.2 mcg/ml y 2.1 mcg/ml, respectivamente. No hay indicios de acumulación del fármaco con dosis repetidas durante 10 días cuando se administra en las dosis recomendadas. Con función renal normal, 91.9 % de una dosis intramuscular se excreta sin cambio en la orina en las primeras 8 horas y 98.2 % dentro de 24 horas. Las concentraciones de orina medias para 6 horas son 563 mcg/ml después de una dosis de 250 mg, 697 mcg/ml después de una dosis de 375 mg y 832 mcg/ml después de una dosis de 500 mg. Los estudios con recién nacidos de diferentes pesos (menos de 1.5 kg, 1.5 a 2.0 kg, sobre 2.0 kg) que recibieron AMIKIN® de forma intramuscular a una dosis de 7.5 mg/kg revelaron que, como es el caso de otros aminoglucósidos, los valores de vida media en suero se correlacionaron de manera inversa con la edad postnatal y las depuraciones renales de la amikacina. El volumen de la distribución indica que la amikacina, como otros aminoglucósidos, permanece principalmente en el espacio del líquido extracelular de los lactantes. La dosificación repetida cada 12 horas en todos los grupos anteriores no demuestra la acumulación después de 5 días. Administración intravenosa: Las dosis únicas de 500 mg (7.5 mg/kg) administradas a adultos normales como una infusión durante un periodo de 30 minutos produjeron una concentración máxima en suero media de 38 mcg/ml al final de la infusión y concentraciones de 24 mcg/ml, 18 mcg/ml y 0.75 mcg/ml a los 30 minutos, 1 hora y 10 horas después de la infusión, respectivamente. Se excretó el 84 % de la dosis administrada en la orina en 9 horas y 94 % dentro de un periodo de 24 horas. Las infusiones de 7.5 mg/kg cada 12 horas en adultos normales fueron bien toleradas y no causaron acumulación del fármaco. Las dosis únicas de 15 mg/kg administradas por vía intravenosa durante 30 minutos a adultos voluntarios con función renal normal produjeron concentraciones en suero máximas medias de 77 mcg/ml y concentraciones de 47 mcg/ml y 1 mcg/ml a las 1 y 12 horas, respectivamente, después de la infusión. Se observó una concentración en suero máxima media de 55 mcg/ml después de una infusión de 30 minutos de 15 mg/kg en pacientes mayores (depuración media de la creatinina de 64 ml/min) con concentraciones en suero de 5.4 mcg/ml a las 12 horas y 1.3 mcg/ml a las 24 horas después de la infusión. En estudios con dosis múltiples no se observó ninguna acumulación en pacientes con función renal normal que recibieron dosis una vez al día de 15 a 20 mg/kg.
Contraindicaciones: AMIKIN® está contraindicado en pacientes con alergia conocida a la amikacina o a cualquier componente de la formulación. Antecedentes de hipersensibilidad o reacciones tóxicas graves a los aminoglucósidos pueden contraindicar el uso de cualquier aminoglucósido debido a las sensibilidades cruzadas conocidas de pacientes a los fármacos de esta clase.
Precauciones generales advertencias y precauciones para el uso: Los pacientes tratados con aminoglucósidos por vía parenteral deberán estar bajo observación clínica cercana debido a la ototoxicidad y nefrotoxicidad potenciales asociadas con su uso. Los periodos de seguridad para el tratamiento por más de 14 días no se han establecido. Se puede presentar neurotoxicidad, manifestada como ototoxicidad vestibular y/o auditiva bilateral, en pacientes tratados con aminoglucósidos. El riesgo de ototoxicidad inducida por aminoglucósidos es mayor en pacientes con insuficiencia renal y en aquellos que reciben altas dosis o en aquellos cuya terapia es prolongada. Por lo general primero se presenta sordera de alta frecuencia y solo se puede detectar mediante pruebas audiométricas. Se puede presentar vértigo y puede ser indicio de lesión vestibular. Otras manifestaciones de neurotoxicidad pueden incluir entumecimiento, hormigueo en piel, contracciones musculares y convulsiones. El riesgo de ototoxicidad debido a aminoglucósidos se incrementa con el grado de exposición a concentraciones en suero, ya sean máximas o mínimas, de manera persistente. Los pacientes que desarrollan daño coclear o vestibular pudieran no presentar síntomas durante la terapia para advertirles que están desarrollando toxicidad del octavo par craneal y sordera bilateral irreversible total o parcial o se puede presentar vértigo discapacitante después de suspender la administración del fármaco. Por lo general, la ototoxicidad inducida por aminoglucósidos es irreversible. Los aminoglucósidos son potencialmente nefrotóxicos. El riesgo de nefrotoxicidad es mayor en pacientes con insuficiencia renal y en aquellos que reciben altas dosis o en aquellos cuya terapia es prolongada. La función renal y del octavo par craneal, deben ser supervisadas cercanamente en especial en pacientes con insuficiencia renal conocida o presunta al inicio de la terapia y también en aquellos cuya función renal sea normal al inicio, pero que comience a desarrollar signos de disfunción renal durante la terapia. Las concentraciones séricas de amikacina deben ser supervisadas cuando sea posible para asegurar las concentraciones adecuadas y para evitar concentraciones tóxicas potenciales. Se debe hacer un examen de orina para evaluar la densidad urinaria, incremento en la excreción de proteínas y la presencia de células o cilindros. Se deben medir de manera periódica el nitrógeno ureico en sangre, creatinina sérica o depuración de la creatinina. Se deben obtener audiogramas en serie cuando sea posible en pacientes suficientemente mayores como para hacerles estas pruebas, en particular pacientes en riesgo alto. Los indicios de ototoxicidad (mareo, vértigo, tinnitus, ruido en los oídos y pérdida de la audición) o nefrotoxicidad requieren la suspensión del fármaco o ajuste de la dosis. Se debe evitar el uso concurrente o secuencial de otros productos neurotóxicos o nefrotóxicos por vía sistémica oral o tópica, en particular bacitracina, cisplatino, amfotericina B, cefaloridina, paromomicina, viomicina, polimixina B, colistina, vancomicina u otros aminoglucósidos. Otros factores que pueden incrementar el riesgo de toxicidad son la edad avanzada y la deshidratación. Se debe evitar el uso concurrente de AMIKIN® con diuréticos potentes (ácido etacrínico o furosemida) dado que los diuréticos por sí mismos pueden causar ototoxicidad. Adicionalmente, cuando se administran por vía intravenosa, los diuréticos pueden aumentar la toxicidad del aminoglucósido al alterar las concentraciones del antibiótico en suero y tejido. Se han registrado bloqueo neuromuscular y parálisis respiratoria después de inyección parenteral, instilación tópica (como en irrigación ortopédica y abdominal o en el tratamiento local de empiema) y después del uso oral de aminoglucósidos. Se debe considerar la posibilidad de parálisis respiratoria si se administran aminoglucósidos por cualquier vía, en especial en pacientes que reciben anestésicos, bloqueadores neuromusculares tales como la tubocurarina, succinilcolina, decametonio o en pacientes que reciben transfusiones masivas de sangre anticoagulada con citrato. Si se presenta bloqueo neuromuscular, las sales de calcio pueden revertir la parálisis respiratoria, pero pudiera necesitarse asistencia respiratoria mecánica. AMIKIN® en viales contiene bisulfito de sodio, un sulfito que puede causar reacciones tipo alérgicas incluidos síntomas anafilácticos y que ponen en peligro la vida o episodios asmáticos menos graves en ciertas personas susceptibles. La prevalencia total de la sensibilidad al sulfito en la población general es poco común y probablemente baja. La sensibilidad al sulfito es más frecuente en sujetos asmáticos que en sujetos no asmáticos. Los aminoglucósidos se absorben rápida y totalmente cuando se administran por vía tópica, excepto por la vejiga urinaria, en asociación con procedimientos quirúrgicos. Se han registrado sordera irreversible, insuficiencia renal y muerte debido a bloqueo neuromuscular después de la irrigación de campos quirúrgicos pequeños y grandes con una preparación de aminoglucósido. Se debe evitar el uso concurrente o en serie de otros agentes ototóxicos o nefrotóxicos por vía sistémica o tópica debido al potencial de efectos aditivos. Se ha registrado un aumento en la nefrotoxicidad después de la administración concomitante parenteral de antibióticos aminoglucósidos y cefalosporinas. El uso concomitante de cefalosporina puede elevar de manera falsa las determinaciones de la concentración en suero de creatinina. Ototoxicidad: (Ver Precauciones generales, advertencias y precauciones para el uso). Nefrotoxicidad: Los pacientes deben estar bien hidratados durante el tratamiento y se debe evaluar la función renal mediante los métodos usuales antes de iniciar la terapia y diariamente durante el curso del tratamiento. Se requiere una reducción de la dosificación (ver Dosis y vía de administración, Dosis recomendada) si se presentan indicios de disfunción renal tales como la presencia de cilindros urinarios, leucocitos o eritrocitos, albuminuria, disminución en la depuración de la creatinina, disminución de la densidad urinaria, incremento del BUN, creatinina sérica u oliguria. Si se incrementa la azoemia o si se presenta disminución progresiva en la eliminación urinaria, se debe suspender el tratamiento. Los pacientes adultos mayores pueden tener una función renal reducida que pudiera no ser evidente en las pruebas de escrutinio de rutina como el BUN o la creatinina sérica. Una determinación de la depuración de creatinina puede ser más útil. La supervisión de la función renal en pacientes adultos mayores durante el tratamiento con aminoglucósidos es de particular importancia. Neurotoxicidad: Se ha demostrado bloqueo neuromuscular y parálisis muscular en animales de laboratorio que recibieron dosis altas de amikacina. Se debe considerar la posibilidad de bloqueo neuromuscular y parálisis respiratoria cuando se administra amikacina de forma concomitante con anestésicos o bloqueadores neuromusculares. Si se presenta un bloqueo, las sales de calcio pueden revertir este fenómeno. Los aminoglucósidos se deben usar con precaución en pacientes con trastornos musculares tales como miastenia grave o parkinsonismo dado que estos fármacos pueden agravar la debilidad muscular debido a su potencial efecto tipo curare sobre la unión neuromuscular. Otros: Como con otros antibióticos, el uso de la amikacina puede provocar el sobrecrecimiento de organismos no susceptibles. Si esto pasa, se debe instituir la terapia adecuada. La mezcla in vitro de aminoglucósidos con antibióticos betalactámicos (penicilinas o cefalosporinas) puede dar como resultado una importante inactivación mutua. También se puede presentar una reducción en la actividad en suero cuando se administra in vivo un aminoglucósido o fármaco de tipo penicilina por vías separadas. La inactivación del aminoglucósido es clínicamente significativa solo en pacientes con insuficiencia renal grave. La inactivación puede continuar en muestras de líquidos corporales obtenidas para el ensayo, lo que produce lecturas del aminoglucósido inexactas. Tales muestras deben manejarse de manera adecuada (analizadas de maneras inmediatas, congeladas o tratadas con betalactamasa). Uso pediátrico: Los aminoglucósidos deben usarse con precaución en niños prematuros o neonatos debido a la inmadurez renal de estos pacientes y la prolongación resultante de la vida media en suero de estos fármacos. Efectos sobre la capacidad para conducir y operar maquinaria: Debido a la aparición de algunas reacciones adversas, la capacidad para conducir y operar maquinaria puede ser alterada (Ver Reacciones secundarias y adversas).
Restricciones de uso durante el embarazo y lactancia: Los aminoglucósidos pueden causar daño fetal cuando se administran a una mujer embarazada. Los aminoglucósidos cruzan la placenta y existen varios informes de sordera congénita total bilateral irreversible en niños cuyas madres recibieron estreptomicina durante el embarazo. Si bien no se han registrado efectos secundarios graves en fetos o recién nacidos en el tratamiento de mujeres embarazadas con otros aminoglucósidos, existe el potencial de daño. Se han llevado a cabo estudios de reproducción con amikacina en ratas y ratones, y no mostraron indicios de afectación de la fertilidad o daño al feto debido a la amikacina. No hay estudios bien controlados en mujeres embarazadas, pero la experiencia de las investigaciones no incluye ninguna evidencia positiva de efectos adversos en el feto. Si se usa este fármaco durante el embarazo o si la paciente queda embarazada mientras está tomando éste fármaco, se debe informar a la paciente sobre el riesgo potencial al feto. Se desconoce si el fármaco se excreta en la leche materna. Como regla general, no se debe amamantar mientras una paciente esté recibiendo algún fármaco, ya que muchos medicamentos son excretados en la leche materna.
Reacciones secundarias y adversas: Todos los aminoglucósidos tienen el potencial de inducir ototoxicidad, toxicidad renal y bloqueo neuromuscular (Ver Precauciones generales, advertencias y precauciones para el uso). Estas toxicidades se presentan con mayor frecuencia en pacientes con insuficiencia renal, en pacientes tratados con otros fármacos ototóxicos o nefrotóxicos y en pacientes tratados durante periodos más largos y/o con dosis más altas que las recomendadas. Neurotoxicidad - ototoxicidad: Los efectos tóxicos sobre el octavo nervio craneal pueden dar como resultado pérdida de la audición, pérdida del equilibrio o ambos. La amikacina afecta principalmente la función auditiva. El daño coclear incluye sordera de altas frecuencias y por lo general se presenta antes de que se pueda detectar la pérdida clínica de la audición mediante pruebas audiométricas. Neurotoxicidad - bloqueo neuromuscular: Se pueden presentar parálisis muscular aguda y apnea después del tratamiento con aminoglucósidos. Nefrotoxicidad: Se puede presentar elevación de la creatinina sérica, albuminuria y presencia de eritrocitos, leucocitos o cilindros en orina, azoemia y oliguria. Los cambios en la función renal son por lo general reversibles cuando se suspende la administración del fármaco. Como se esperaría con cualquier aminoglucósido, se han recibido informes de nefropatía tóxica e insuficiencia renal aguda durante la vigilancia posterior a la comercialización. Otras: Otras reacciones adversas que se han registrado en raras ocasiones incluyen exantema cutáneo, fiebre medicamentosa, cefalea, parestesias, temblor, náusea y vómito, eosinofilia, artralgia, anemia, hipotensión e hipomagnesemia. Se ha registrado respuesta anafiláctica (reacción anafiláctica, choque anafiláctico y reacción anafilactoide), broncoespasmo, hipersensibilidad, prurito y urticaria durante la vigilancia posterior a la comercialización. Se ha registrado infarto macular que algunas veces lleva a la pérdida permanente de la visión después de la administración intravítrea (inyección en el ojo) de amikacina. Experiencia clínica: (Ver Reacciones secundarias y adversas). Experiencia postcomercialización: (Ver Reacciones secundarias y adversas).
Interacciones medicamentosas y de otro género: Existe un aumento en el riesgo de hipocalcemia cuando se administran aminoglucósidos con biofosfonatos. Existe un aumento en el riesgo de nefrotoxicidad y posiblemente de ototoxicidad cuando se administran aminoglucósidos con componentes de platino. La indometacina puede incrementar las concentraciones plasmáticas de amikacina en los neonatos. La tiamina (vitamina B1) administrada de forma concomitante, puede ser destruida por el componente reactivo bisulfito de sodio de la formulación de sulfato de amikacina.
Alteraciones en los resultados de pruebas de laboratorio: (Ver Reacciones secundarias y adversas).
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: No se han llevado a cabo estudios a largo plazo en animales para evaluar el potencial carcinogénico y no se ha estudiado la mutagenicidad. AMIKIN® administrado a ratas a dosis hasta por 10 veces la dosis diaria en humanos, no alteraron la fertilidad masculina o femenina.
Dosis y vía de administración: Vía de administración: Intramuscular o intravenosa (venoclisis) dosificación recomendada: Se debe obtener el peso corporal del paciente previo al tratamiento para el cálculo de la dosificación correcta. AMIKIN® puede administrarse por vía intramuscular o intravenosa. Se debe estimar el estado de la función renal con la medición de la concentración de la creatinina sérica o el cálculo de la tasa de depuración endógena de la creatinina. El nitrógeno ureico en sangre (BUN) es mucho menos confiable para este propósito. Se debe hacer la reevaluación de la función renal de manera periódica durante la terapia. Cuando sea posible, se deberán medir las concentraciones de amikacina en suero para asegurar concentraciones adecuadas pero no excesivas. Es conveniente medir las concentraciones séricas máxima y mínima de manera intermitente durante la terapia. Se deben evitar las concentraciones máximas (30-90 minutos después de la inyección) por arriba de 35 mcg/ml y las concentraciones mínimas (justo antes de la siguiente dosis) por arriba de 10 mcg/ml. Se debe ajustar la dosificación como se indica. En pacientes con función renal normal, se puede usar una dosificación de una vez al día; las concentraciones máximas en estos casos puede exceder los 35 mcg/ml. (Ver Dosis y vía de administración, Dosificación una vez al día y Dosis y vía de administración, Insuficiencia renal a continuación). Administración intramuscular e intravenosa en pacientes con función renal normal: La dosis intramuscular o intravenosa recomendada para adultos, niños y lactantes de mayor edad con función renal normal es de 15 mg/kg/día dividida en 2 o 3 dosis iguales administradas en intervalos divididos de manera equitativa; es decir, 7.5 mg/kg cada 12 horas o 5 mg/kg cada 8 horas. El tratamiento de pacientes con gran masa corporal no debe exceder de 1.5 g/día. En prematuros, la dosis recomendada es 7.5 mg/kg cada 12 horas. Los recién nacidos deben recibir 10 mg/kg como dosis inicial y 7.5 mg/kg cada 12 horas de ahí en adelante. Los lactantes mayores de 2 semanas y los niños deben recibir 7.5 mg/kg cada 12 horas o 5 mg/kg cada 8 horas como se especificó anteriormente. Dosificación una vez al día: De manera alterna, se puede considerar en pacientes con función renal normal reflejada por depuración de la creatinina ≥50 ml/min., una dosis diaria única intravenosa de 15 mg/kg/día en adultos o 20 mg/kg/día en niños (4 semanas o mayores) para bacteriemia, septicemia, infecciones del aparato respiratorio, infecciones de las vías urinarias complicadas, infecciones intrabdominales y uso empírico en neutropenia febril. La información sobre el uso de la dosificación de una vez al día en pacientes con involucro de otros órganos es limitada. (Ver también lo anterior respecto al monitoreo de las concentraciones en suero máximas y mínimas de amikacina). Cuando AMIKIN® está indicado para infecciones no complicadas de las vías urinarias, se puede administrar una dosis diaria de 500 mg ya sea como dosis única o en 2 dosis divididas (250 mg 2 veces al día). Se debe tener cuidado al calcular la dosis de manera exacta y se debe diluir más la solución reconstituida de 50 mg/ml cuando sea necesario para permitir la administración de dosis exactas en el prematuro. La duración usual del tratamiento es de 7 a 10 días. La dosis diaria total por todas las vías de administración no debe exceder de 15-20 mg/kg/día. En infecciones difíciles y complicadas donde se considere el tratamiento más allá de 10 días, se debe reevaluar el uso de AMIKIN® y si se continúa, se deberá supervisar la función renal, auditiva y vestibular, así como la concentración de amikacina en suero. En las dosis recomendadas, las infecciones no complicadas debidas a organismos sensibles a la amikacina deben responder en 24 a 48 horas. Si la respuesta clínica definitiva no se presenta dentro de 3 a 5 días, se debe suspender la terapia y se debe volver a verificar el patrón de susceptibilidad al antibiótico del organismo invasor. Si la infección no cede puede deberse a la resistencia del organismo o a la presencia de focos sépticos que requieren drenaje quirúrgico. Insuficiencia renal: NOTA: En pacientes con insuficiencia renal reflejada por depuración de creatinina < 50 ml/min., no es aconsejable la administración de la dosis diaria total recomendada de amikacina en una dosis diaria única ya que estos pacientes tendrán una exposición prolongada a concentraciones mínimas altas. Véase a continuación para los ajustes de la dosificación en pacientes con insuficiencia renal. Para pacientes con insuficiencia renal que reciben una dosificación diaria de 2 o 3 veces al día, cuando sea posible, se deben supervisar las concentraciones séricas de amikacina mediante el procedimiento adecuado del ensayo. Se deben ajustar las dosis en pacientes con insuficiencia renal ya sea al administrar la dosis normal en intervalos prolongados o al administrar dosis reducidas en intervalos fijos. Ambos métodos se basan en los valores de depuración de creatinina o en los valores de creatinina sérica del paciente ya que se ha encontrado que correlacionan con la vida media del aminoglucósido en pacientes con función renal disminuida. Estos programas de dosificación se deben usar en conjunto con observaciones clínicas y de laboratorio del paciente y se deben modificar conforme sea necesario, incluida la modificación cuando se lleve a cabo diálisis. Dosis normal en intervalos prolongados entre las dosis: Si la tasa de depuración de la creatinina no está disponible y la condición del paciente es estable, se puede calcular un intervalo de dosificación en horas para la dosis normal única (es decir, la que se administraría a pacientes con función renal normal en un programa BID, 7.5 mg/kg) al multiplicar la creatinina sérica del paciente por 9; por ejemplo, si la concentración de creatinina sérica es 2 mg/100 ml, la dosis única recomendada (7.5 mg/kg) se deberá administrar cada 18 horas. Dosis reducidas en intervalos de tiempo fijos entre dosificaciones: Cuando la función renal está alterada y es conveniente administrar AMIKIN® en un intervalo de tiempo fijo, se debe reducir las dosis. En estos pacientes, se deben medir las concentraciones en suero de amikacina para asegurar la administración exacta y para evitar concentraciones séricas excesivas. Si no están disponibles las determinaciones séricas del ensayo y la condición del paciente es estable, los valores de creatinina sérica y depuración de la creatinina son indicadores del grado de insuficiencia renal que se deben usar como guía para la dosificación. Primero, inicie la terapia al administrar una dosis normal, 7.5 mg/kg, como dosis inicial. Esta dosis es la misma que la dosis normalmente recomendada que se calcula para un paciente con una función renal normal como se describió anteriormente. Para determinar el tamaño de la dosis de mantenimiento administrada cada 12 horas, se debe reducir la dosis inicial en proporción con la reducción en la tasa de depuración de la creatinina del paciente: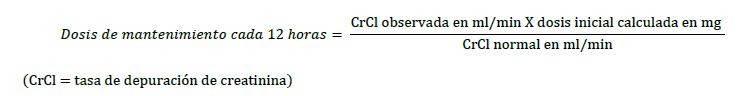
Una guía aproximada alterna para determinar la dosificación reducida a intervalos de 12 horas (para pacientes cuyos valores de creatinina sérica en estado estable se conocen) es dividir la dosis normalmente recomendada entre la creatinina sérica del paciente. Los programas de dosificación anteriores no pretenden ser recomendaciones rígidas, pero se proporcionan como guías para la dosificación cuando la medición de las concentraciones en suero de la amikacina no es posible. Pediátricos y adolescentes: (Ver Dosis y vía de administración, dosis recomendada). Instrucciones especiales para el uso, manejo y disposición: Se deben inspeccionar de manera visual los medicamentos parenterales respecto a partículas en suspensión y decoloración antes de la administración si la solución y el envase lo permiten. Los aminoglucósidos administrados por cualquier vía no deben premezclarse con otros medicamentos sino que se deben administrar de forma separada. Debido a la potencial toxicidad de los aminoglucósidos, no se aconsejan las recomendaciones de "dosificación fija" que no se basen en el peso corporal. En su lugar, es importante calcular la dosificación para que cubra las necesidades de cada paciente. Administración intravenosa: preparación de las soluciones: La solución para uso intravenoso se prepara al agregar la dosis deseada a 100 ml o 200 ml de diluyente estéril tal como solución salina o dextrosa al 5 % en agua o cualquier otra solución compatible. La solución se administra a adultos en periodos de 30 a 60 minutos. La dosis diaria total no debe exceder de 15-20 mg/kg/día. En pacientes pediátricos, la cantidad de líquido usado dependerá de la cantidad tolerada por el paciente. Deberá haber una cantidad suficiente para infundir la amikacina durante un periodo de 30 a 60 minutos. Los lactantes deben recibir una infusión de 1 a 2 horas. La amikacina no debe ser premezclada físicamente con otros medicamentos, sino que se debe administrar de manera separada de acuerdo con la dosis y vía recomendada. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: En el caso de sobredosis o reacción tóxica, la diálisis peritoneal o hemodiálisis será una ayuda en la eliminación de la amikacina de la sangre. También se pueden reducir las concentraciones de amikacina durante hemofiltración arteriovenosa continua. En el recién nacido, también se puede considerar la exsanguineotransfusión.
Presentaciones: AMIKIN inyectable 1 gr. Caja con 1 ampolleta o 1 frasco ámpula de 4 ml. AMIKIN inyectable 100 mg (Ped) Caja con 2 ampolletas o 1 o 2 frascos ámpula de 2 ml. AMIKIN inyectable 250 mg Caja con 2 ampolletas o 1 o 2 frascos ámpula de 2 ml. AMIKIN inyectable 500 mg Caja con 2 ampolletas o 1 o 2 frascos ámpula de 2 ml.
Recomendaciones sobre almacenamiento: Consérvese a no más de 30°C. En ocasiones la solución puede tomar una coloración amarilla muy pálida, sin embargo esto no indica una disminución de su potencia. Estabilidad en líquidos IV: AMIKIN® es estable durante 24 horas a temperatura ambiental a concentraciones de 0.25 y 5.0 mg/ml en las siguientes soluciones: Solución de dextrosa al 5 %: Solución de dextrosa al 5 % y cloruro de sodio al 0.2 %. Solución de dextrosa al 5 % y cloruro de sodio 0.45 %. Solución de cloruro de sodio al 0.9 % Solución de Ringer lactada: Normosol® M en inyección de dextrosa al 5 % (o inyección Plasma-Lyte 56 en dextrosa al 5 % en agua) Normosol® R en inyección de dextrosa al 5 % (o inyección Plasma-Lyte 148 en dextrosa al 5 % en agua). En las anteriores soluciones con AMIKIN® en concentraciones de 0.25 y 5.0 mg/ml, las soluciones conservadas durante 60 días a 4 °C y luego almacenadas a 25 °C tienen tiempos de uso de 24 horas.
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre durante el embarazo y la lactancia. Reporte las sospechas de reacción adversa a los correos: Farmacovigilancia COFEPRIS: farmacovigilancia@cofepris.gob.mx.
Nombre y domicilio del laboratorio: Hecho en Italia por: Bristol-Myers Squibb S.R.L. Loc. Fontana del Ceraso, 03012 Anagni (FR), Italia. Para: Bristol-Myers Squibb Manufacturing Company. Road 3, Km 77.5, Post Box 609, Humacao, PR-00791, EUA. Importado y Distribuido por: Bristol-Myers Squibb de México, S. de R.L. de C.V. Rancho 4 Milpas km 1 Carretera Tepotzotlán - La Aurora, MDC Fase II, Sección "D", Col. Ex Hacienda San Miguel, C.P. 54715, Cuautitlán Izcalli, México. Representante legal: Bristol-Myers Squibb de México, S. de R.L. de C.V. Avenida Insurgentes Sur No.1602, Piso 5, Col. Crédito Constructor, C.P. 03940, Deleg. Benito Juárez, Distrito Federal, México.
Número de registro del medicamento: 86876 SSA. Fecha de CCDS: CCDS Amikacin Sulfate Injection, 18 April 2012. Fecha de aprobación registro sanitario: 9 Jul 2015.