CERVARIX®
GSK
Denominación genérica: Vacuna recombinante contra el Virus del Papiloma Humano (Proteína L1).
Forma farmacéutica y formulación: Suspensión inyectable. El frasco ámpula o jeringa prellenada con 1 dosis de 0.5 mL contiene: Proteína L1 del virus del papiloma humano tipo 161: 20 mg. Proteína L1 del virus del papiloma humano tipo 181: 20 mg. 3-O-desacil-4'- monofosforil lípido A (MPL) 2: 50 mg. Hidróxido de aluminio hidratado2: 0.5 mg: Al3+.1Proteína L1 en la forma de partículas no infecciosas semejantes al virus (VLP) producida mediante tecnología de ADN recombinante usando un sistema de expresión de baculovirus. 2El sistema adyuvante AS04, patentado por GlaxoSmithKline, está compuesto de hidróxido de aluminio y 3-O-desacil-4'-monofosforil lípido A (MPL) (ver Farmacodinamia). Lista de excipientes: Cloruro de sodio, fosfato de sodio dihidrogenado dihidratado, agua para inyectables.
Indicaciones terapéuticas: Cervarix® está indicada desde los 9 años de edad en adelante para la prevención de infección persistente, de lesiones anogenitales premalignas (de cérvix, vulvares, vaginales y anales), y de cáncer de cérvix, vulvar, vaginal y anal (carcinoma de células escamosas y adenocarcinoma) causados por Virus del Papiloma Humano (VPH) oncogénicos (ver Advertencias y precauciones y Farmacodinamia).
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: Grupo farmacoterapéutico: Vacunas contra el virus del papiloma, J07BM02. Mecanismo de acción: Se ha demostrado que la infección persistente con tipos oncogénicos de VPH es responsable de casi todos los casos de cáncer cérvico-uterino en el mundo. Cervarix® es una vacuna recombinante no infecciosa preparada usando las partículas similares al virus (VLP) altamente purificadas de la proteína principal L1 de la cápside de los VPH oncogénicos tipos 16 y 18. Ya que las VLP no contienen ADN viral, no pueden infectar las células, reproducirse u ocasionar enfermedades. En estudios animales se ha demostrado que la eficacia de las vacunas basadas en VLP de L1 está mediada principalmente por el desarrollo de una respuesta inmune humoral y memoria inmune mediada por células. Cervarix® tiene un adyuvante AS04, el que en estudios clínicos ha demostrado que induce una respuesta inmune superior y de mayor duración que la inducida por los mismos antígenos conteniendo sólo sal de aluminio [Al(OH)3] como adyuvante. El cáncer de cérvix invasivo incluye al carcinoma cervical escamoso (84%) y al adenocarcinoma (16%, hasta 20% en los países desarrollados con programas de tamizaje). El VPH-16 y el VPH-18 son responsables de aproximadamente el 70% de los casos de cáncer de cérvix, el 80% de los casos del cáncer vulvar y vaginal, el 90% de los casos de cáncer anal, el 70% de los casos de neoplasia intraepitelial de alto grado vulvar (NIV 2/3) y vaginal (NIVa 2/3) y del 78% de los casos de neoplasia intraepitelial de alto grado anal (NIA 2/3) relacionados con VPH en todas las regiones del mundo. Otros tipos oncogénicos del VPH (VPH-31, -33, -35, -39, -45, -51, -52, -56, -58, -59, -66, -68) pueden también causar cánceres anogenitales. Los 4 tipos más comunes de VPH identificados en el carcinoma cervical escamoso (aproximadamente 76%) y en el adenocarcinoma (aproximadamente 91%) son los VPH-16, -18, -45 y -31. Evidencia de Respuesta Anamnésica (Memoria Inmune): La administración de una dosis adicional de la vacuna tras una media de 6.8 años después de la primera vacunación provocó una respuesta inmune anamnésica frente a VPH-16 y VPH-18 (por ELISA y el ensayo de neutralización basado en pseudovirión) en el día 7. Un mes después de esta dosis de exposición, los títulos medios geométricos medios (TMG) excedieron los observados un mes después del esquema de vacunación primaria. Con ELISA, también se observó una respuesta anamnésica para los tipos relacionados- VPH-31 y VPH-45. Eficacia profiláctica: Eficacia clínica en mujeres de 15 a 25 años: Se evaluó la eficacia de Cervarix® en 2 estudios clínicos controlados, doble ciego, aleatorizados (HPV-001/007 y HPV-008) donde se incluyeron un total de 19,778 mujeres con edad entre 15 y 25 años al momento de ser incluidas. El estudio clínico HPV-001/007 se hizo en Norteamérica y Latinoamérica. El estudio HPV- 023 siguió a los sujetos de la cohorte brasileña del estudio 001/007. Los criterios de inclusión en el estudio fueron: mujeres con prueba negativa al ADN de VPH oncogénico (VPH-16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66 y 68) en muestras del cuello uterino, seronegativas para anticuerpos contra VPH-16 y VPH-18 y citología normal. Estas características son representativas de una población de mujeres que se puede suponer no han tenido infección previa por los tipos oncogénicos del VPH antes de la vacunación. El estudio clínico HPV-008 se realizó en Norteamérica, Latinoamérica, Europa, Asia Pacífico y Australia. Se obtuvieron muestras antes de la vacunación para pruebas de ADN de VPH oncogénicos (VPH-16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66 y 68) y pruebas serológicas de anticuerpos contra VPH-16 y VPH-18. Las mujeres fueron vacunadas independientemente de la citología inicial, del estatus serológico a VPH y del ADN viral. Estas características son representativas de una población que incluye mujeres con evidencia de infección previa y/o actual por VPH. Como es el caso en cualquier estudio de eficacia profiláctica, las participantes infectadas inicialmente por un tipo particular de VPH no fueron elegibles para la evaluación de eficacia correspondiente a ese tipo. En los estudios clínicos se usó como un marcador subrogado para cáncer de cérvix a las lesiones llamadas neoplasia intraepitelial cervical (NIC) de grado 2 y 3 (NIC2+). Se ha demostrado también que la infección persistente que dura por lo menos 6 meses es un marcador subrogado relevante para cáncer de cérvix. Aunque la NIC de grado 1 no es un marcador subrogado para cáncer de cérvix, estas lesiones requieren seguimiento médico. 1. Eficacia de la vacuna contra VPH-16/18 en mujeres sin infección previa por tipos oncogénicos de VPH (estudios HPV-001/007/023): En la Tabla 1 se presentan los resultados de eficacia para variables de desenlace histológicas asociadas con VPH-16 y/o VPH-18 (VPH-16/18) que se observaron en el estudio HPV-001/007 (Cohorte Total, es decir, mujeres que recibieron al menos una dosis de la vacuna).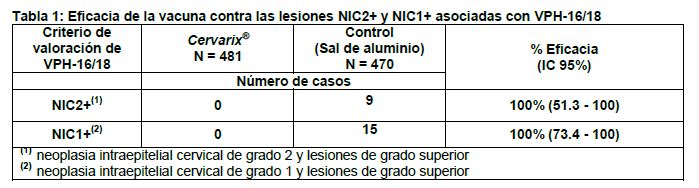
La eficacia contra las anormalidades citológicas causadas por VPH-16/18 fue 96.7% (IC 95%: 87.3-99.6). La eficacia contra la infección persistente por VPH-16/18 fue 98.2% (IC 95%: 89.5- 100) y 96.9% (IC 95%: 81.4-99.9) utilizando una definición de 6 meses y 12 meses, respectivamente. En el estudio HPV-023, los sujetos (N=437) se siguieron por 9.4 años (aproximadamente 113 meses) después de la primera dosis. No hubo ningún nuevo caso de infección o lesiones histopatológicas asociadas con los tipos de VPH 16/18 en el grupo vacunado. En el grupo placebo, hubo 4 casos de infección persistente a 6 meses, un caso de infección persistente a 12 meses y un caso de NIC1+ asociado a VPH 16/18. En el análisis descriptivo combinado de los estudios HPV 001/007/023, la eficacia contra infecciones incidentes e infecciones persistentes por 6 meses por VPH 16/18 fue de 91% (IC 95%: 80.2-96.5) y 96.8% (IC 95%: 80.4-99.9), respectivamente. A pesar de la evidencia de una exposición continua a infecciones por VPH como se observa en el grupo de control, no hay datos de disminución de la protección en las mujeres vacunadas. 2. Eficacia de la vacuna en mujeres con evidencia de infección previa y/o actual por VPH (estudio HPV-008): 2.1 Eficacia profiláctica contra VPH-16/18 en mujeres sin infección previa por VPH-16 y/o VPH-18: En el estudio HPV-008, los análisis primarios de eficacia se efectuaron en la cohorte de Acuerdo al Protocolo (cohorte ATP, (According to Protocol): que incluyó a las mujeres que recibieron 3 dosis de la vacuna y que no habían tenido evidencia de infección previa por el tipo relevante de VPH en el mes 0 ni en el mes 6) y la Cohorte Total Vacunada 1 (cohorte TVC-1 (Total Vaccinated Cohort-1): que incluyó a las mujeres que recibieron al menos una dosis de la vacuna y que no habían tenido infección previa por el tipo relevante del VPH en el mes 0). Ambas cohortes incluían mujeres con una citología inicial normal o con lesiones de bajo grado y sólo excluían a mujeres con una citología con lesiones de alto grado (0.5%). Además, se efectuaron análisis de eficacia en la Cohorte Total Vacunada (TVC) más amplia y en el grupo sin infección previa de la TVC. En el estudio HPV-008, aproximadamente un 26% de las mujeres tuvieron evidencia de infección actual y/o previa por VPH-16/18 y menos del 1% de las mujeres, al inicio, tuvieron prueba positiva para ADN de ambos tipos de VPH (VPH 16 y 18). El análisis final del estudio HPV-008 fue desencadenado en función de los eventos, es decir que se realizó cuando se acumularon al menos 36 casos de NIC2+ asociados con VPH-16/18 en la cohorte ATP. La media de duración del seguimiento fue de aproximadamente 39 meses después de la dosis uno. El análisis de fin del estudio se realizó al final del período de seguimiento de 4 años (es decir, 48 meses después de la dosis uno) e incluyó a todos los sujetos de la Cohorte Total Vacunada (TVC). En el análisis especificado por el protocolo, la eficacia de la vacuna contra las NIC1+ y NIC2+ asociadas con VPH-16/18 fue estadísticamente significativa en las cohortes ATP y TVC-1. Investigaciones posteriores identificaron que varios casos de NIC1+, NIC2+ y de NIC3+ incluían varios tipos oncogénicos del VPH en la misma lesión. Con el objeto de distinguir el(los) tipo(s) de VPH al(los) que más probablemente se debía una lesión, del (de los) tipo(s) sólo temporalmente asociado(s), se aplicó una asignación del tipo de VPH (análisis exploratorio). La asignación del tipo de VPH consideró los tipos de VPH detectados por la Reacción en Cadena de la Polimerasa (PCR) en al menos una de las dos muestras citológicas precedentes, además de los tipos detectados en la lesión. Con base en esta asignación del tipo de VPH, el análisis excluyó los casos (en el grupo de la vacuna y en el grupo de control) que no se consideraron que estuviesen causalmente asociados con las infecciones por VPH-16 o VPH-18 adquiridas durante el estudio. En la Tabla 2 se presentan los resultados observados en ambos análisis (análisis especificado por el protocolo y por asignación del tipo de VPH).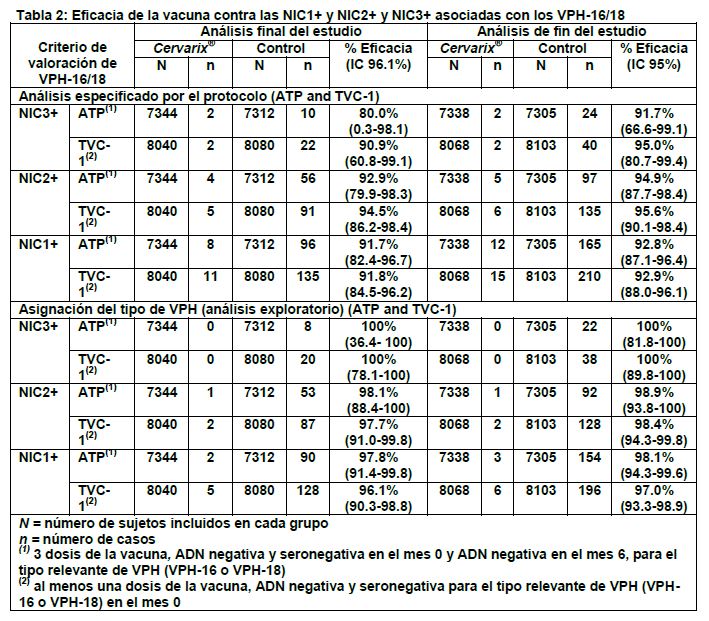
Además, en el momento del análisis final del estudio, se demostró que la eficacia de la vacuna es estadísticamente significativa contra las lesiones NIC2+ asociadas con VPH-16 y VPH-18, individualmente, en ambas cohortes correspondientes a cada análisis. Asimismo se valoró la eficacia de la vacuna contra la infección persistente de 6 meses y de 12 meses, y las anomalías citológicas (≥ASCUS) asociadas con VPH-16/18. La eficacia observada de la vacuna frente a cada criterio de valoración fue estadísticamente significativa en ambas cohortes: En el momento del análisis final del estudio: Infección persistente de 6 meses: 94.3% (91.5;96.3) en la cohorte ATP y 90.2% (87.3-92.6) en la cohorte TVC-1, Infección persistente de 12 meses: 91.4% (86.1-95.0) en la cohorte ATP y 85.3% (79.9- 89.4) en la cohorte TVC-1, Anomalías citológicas (≥ASCUS): 89.0% (84.9-92.1) en la cohorte ATP y 86.7% (82.8- 89.8) en la cohorte TVC-1. En análisis de fin del estudio: Infección persistente de 6 meses: 94.3% (92.0-96.1) en la cohorte ATP y 91.0% (88.5-93.0) en la cohorte TVC-1, Infección persistente de 12 meses: 92.9% (89.4-95.4) en la cohorte ATP y 88.2% (84.5%- 91.2%) en la cohorte TVC-1, Anomalías citológicas (≥ASCUS): 90.7% (87.8-93.1) en la cohorte ATP y 88.6% (85.6- 91.0) en la cohorte TVC-1. En el momento del análisis final del estudio, se observó igualmente que la eficacia de la vacuna es estadísticamente significativa contra las lesiones NIV1+ (neoplasia intraepitelial vulvar de grado 1 y lesiones de grado superior) o las NIVa1+ (neoplasia intraepitelial vaginal de grado 1 y lesiones de grado superior) asociadas con VPH-16/18 en ambas cohortes: 80.0% (IC 96.1%: 0.3-98.1) en la cohorte ATP y 83.2% (IC 96.1%: 20.2-98.4) en la cohorte TVC-1. En el análisis de fin del estudio, la eficacia de la vacuna contra las NIV1+ o NIVa1+ asociadas con VPH-16/18 fue del 75.1% (IC 95%: 22.9-94.0) en la cohorte ATP y del 77.7% (IC 95%: 32. 4-94.5) en la cohorte TVC-1. Hubo 2 casos de NIV2+ o NIVa2+ asociados con VPH-16 o VPH-18 en el grupo de la vacuna y 7 casos en el grupo de control en la cohorte ATP. El estudio no se diseñó con la potencia suficiente como para demostrar una diferencia entre la vacuna y el grupo de control para estos criterios de valoración. No se observó evidencia de protección frente a la enfermedad causada por los tipos de VPH para los que los sujetos eran ADN positivos al inicio del estudio. Sin embargo, los individuos ya infectados antes de la vacunación por uno de los tipos de VPH relacionados con la vacuna se vieron protegidos contra la enfermedad clínica causada por el otro tipo de VPH de la vacuna. 2.2 Impacto global de la vacuna en la carga de la enfermedad por VPH: En el estudio HPV-008 se evaluó la eficacia global de la vacuna independientemente del tipo de ADN de VPH en la lesión y estratificada de acuerdo al estatus de ADN de VPH y al estatus serológico iniciales. En la cohorte TVC y en la cohorte sin infección previa de la TVC, que incluyeron a todas las mujeres vacunadas, se demostró la eficacia de la vacuna contra las NIC3+, NIC2+ y NIC1+ (Tabla 3). Se demostró también el impacto de Cervarix® en la reducción de la terapia cervical local (procedimiento de electroexcisión con asa (LEEP), conización, bisturí o láser) en las mismas cohortes (Tabla 3). El grupo sin infección previa de la TVC es un subconjunto de la TVC que incluye mujeres con citología normal, y que eran ADN de VPH negativas para 14 tipos oncogénicos del VPH (VPH-16, -18, -31, -33, -35, -39, -45, -51, -52, -56, -58, -59, -66, -68) y seronegativas para VPH-16 y VPH-18 al inicio del estudio.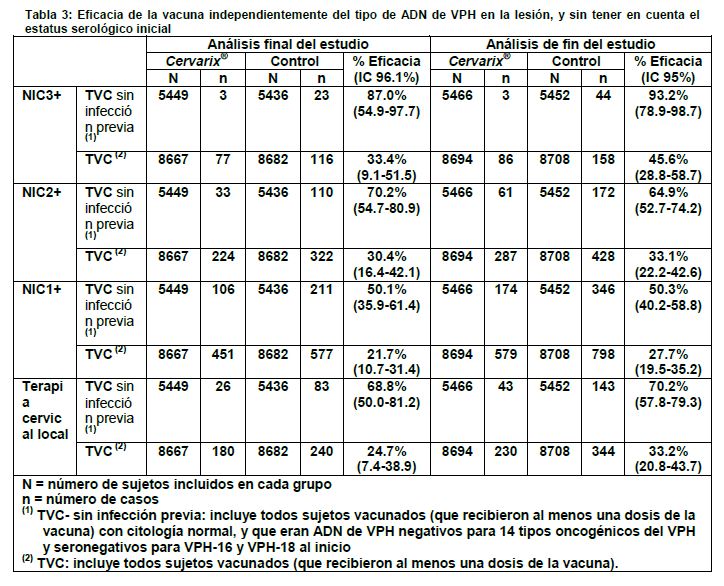
2.3 Eficacia profiláctica contra infección por tipos de VPH oncogénicos diferentes a VPH-16 y a VPH-18: En el estudio HPV-008, se evaluó la eficacia de la vacuna frente a 12 tipos oncogénicos de VPH no incluidos en la formulación de la vacuna (VPH-31, -33, -35, -39, -45, -51, -52, - 56, -58, -59, -66, -68) en las cohortes ATP y TVC-1. En el momento del análisis final del estudio, la eficacia estadísticamente significativa de la vacuna contra las lesiones NIC2+ para todos los tipos combinados del VPH (VPH-31, -33, -35, -39, -45, -51, -52, -56, -58, -59, -66, -68), sin incluir los tipos 16 y 18 del VPH, se demostró con 54.0% (IC 96.1%: 34.0-68.4) en la cohorte ATP y de 46.0% (IC 96.1%: 27.0- 60.3) en la cohorte TVC-1. En el análisis de fin del estudio, la eficacia de la vacuna contra las NIC2+ para todos los tipos de VPH combinados, excluidos los tipos 16 y 18 de VPH, fue del 46.8% (IC 95%: 30.7-59.4) en la cohorte ATP y del 40.8% (IC 95%: 25.5-53.1) en la cohorte TVC-1. En el momento del análisis final del estudio, se ha observado una eficacia estadísticamente significativa de la vacuna frente a la infección persistente de seis meses y frente a las lesiones NIC2+ para los siguientes tipos de VPH individuales: infección persistente de 6 meses: tipos 31, 33, 45 en la cohorte ATP; tipos 31, 33, 45, 51 en la cohorte TVC-1. NIC2+: tipos 31, 51, 58 en la cohorte ATP; tipos 31, 33, 35, 51 en la cohorte TVC-1. En el análisis de fin del estudio, se acumularon más casos y se observó un límite inferior del IC 95% por encima de cero para los tipos 31, 33, 45 y 51 de VPH, tanto para la infección persistente de 6 meses como para las NIC2+ en las cohortes ATP y TVC-1. Para las NIC2+, también se observó un límite inferior del IC 95% por encima de cero para el tipo 39 de VPH en la cohorte ATP y para el tipo 66 de VPH en la cohorte TVC-1. Eficacia clínica en mujeres de 26 años o más: La eficacia de Cervarix® se evaluó en un estudio clínico doble ciego, aleatorizado, de fase III (HPV-015), que incluyó un total de 5777 mujeres de 26 años o más. El estudio se llevó a cabo en América del Norte, América Latina, Pacífico asiático y Europa, y permitió la inclusión de mujeres con antecedentes de enfermedad/infección por VPH. Se efectuó un análisis intermedio cuando todos los sujetos habían completado la visita del mes 48 del estudio. Los análisis primarios de eficacia se efectuaron en la cohorte ATP para la eficacia y en la cohorte TVC. En la siguiente tabla, se resume la eficacia de la vacuna contra el criterio de valoración primario combinado (infección persistente de 6 meses y/o NIC1+) asociado con VPH-16/18.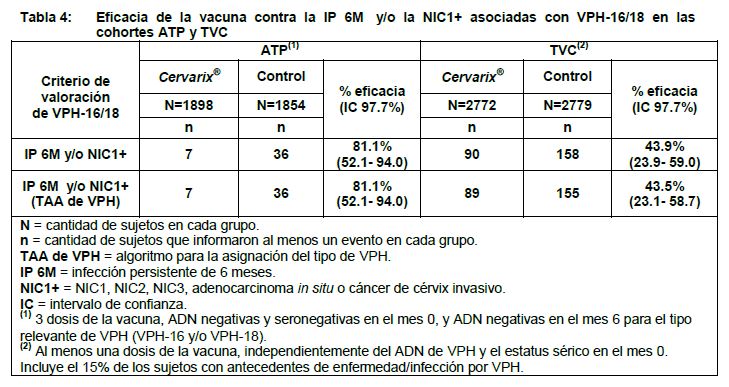
La eficacia de la vacuna contra la infección persistente de 6 meses fue del 79.1% (IC 97.7% [27.6 - 95.9]) para VPH-31 y del 76.9% (IC 97.7% [18.5 - 95.6]) para VPH-45 en la cohorte ATP. Eficacia clínica contra infecciones anales prevalentes en mujeres de 18-25 años de edad: El estudio HPV-009 evaluó la eficacia de la vacuna contra infecciones anales prevalentes en la visita del estudio de 4 años. La eficacia de la vacuna contra VPH-16/18 y contra los tipos de VPH-31/33/45 no vacunales se presenta en la Tabla 5. Se evaluó la infección cervical en las mismas mujeres durante la misma visita para fines de comparación.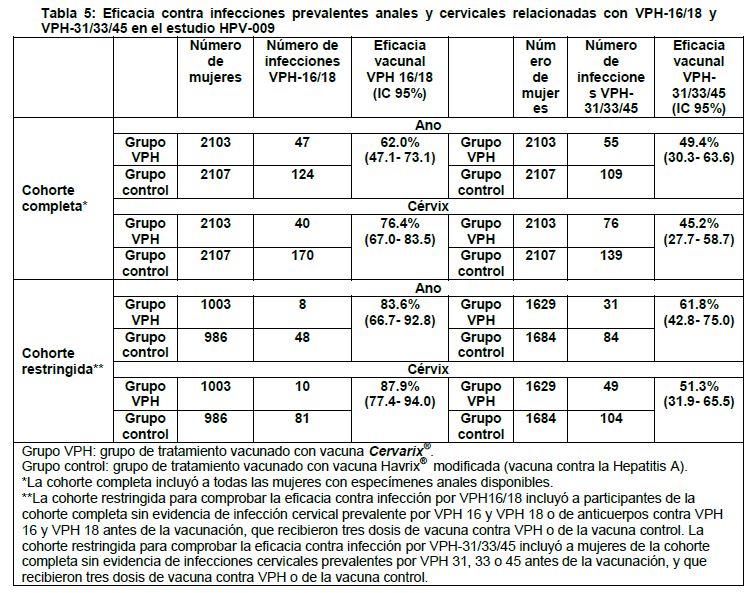
Inmunogenicidad inducida por la vacuna: La respuesta de anticuerpos a VPH-16 y VPH-18 se determinó usando un ELISA de tipo específico que se ha demostrado está correlacionado estrechamente con los análisis de neutralización (incluyendo el ensayo de neutralización basado en pseudovirión desarrollado por el Instituto Nacional de Cáncer de los E.U.). En estudios clínicos se ha demostrado la transudación de anticuerpos del suero a la mucosa del cérvix del útero. Se ha evaluado la inmunogenicidad inducida por las tres dosis de Cervarix® en más de 5000 mujeres entre los 9 y 55 años y en más de 800 varones entre los 10 y 18 años. En estudios clínicos, más del 99% de las participantes inicialmente seronegativas habían seroconvertido a los VPH tipo 16 y 18 un mes después de la tercera dosis. Los títulos medios geométricos (TMG) de IgG inducidos por la vacuna están muy por encima de los títulos observados en mujeres infectadas previamente, pero que habían depurado la infección por VPH (infección natural). Las mujeres que eran inicialmente seropositivas así como las mujeres seronegativas alcanzaron títulos similares después de la vacunación. Inmunogenicidad en mujeres entre 15 y 25 años: En el estudio HPV-001/007 se evaluó la respuesta inmune contra VPH-16 y VPH-18 hasta 76 meses después de la primera dosis, en mujeres con edades entre 15 y 25 años al momento de ser vacunadas. En el estudio HPV-023, esta respuesta inmune continuó siendo evaluada hasta 9.4 años después de la dosis uno, en un subconjunto de la población del estudio HPV-001/007. En el estudio HPV-023, 100% de las mujeres vacunadas fueron seropositivas tanto para VPH-16 como VPH-18 por el método de ELISA o por el ensayo neutralización basado en pseudovirión (PBNA, por su sigla en inglés) hasta 9.4 años después de la primera dosis de la vacuna. Los títulos medios geométricos (TMG) de IgG inducidos por la vacuna para VPH-16 y VPH-18 llegaron al máximo al mes 7 y entonces disminuyeron hasta llegar a un nivel estable a partir del mes 18, sin observarse ninguna disminución sustancial adicional hasta el final del período de seguimiento (mes 113).En el mes 113, los TMG para VPH-16 y VPH-18 continuaban siendo al menos 10 veces más altos que los títulos observados en mujeres infectadas previamente, pero que habían depurado la infección por VPH (infección natural) y el 100% de las mujeres resultaron seropositivas para ambos antígenos. En el estudio HPV-008, la inmunogenicidad hasta el mes 48 fue similar a la respuesta observada en el estudio HPV-001/007. Se observó un perfil cinético similar con los anticuerpos neutralizantes. Se extiende la demostración de la eficacia de Cervarix® en mujeres de 15 a 25 años a otros grupos de edad: En un análisis combinado (HPV-029, -030 y -048), 99.7% y 100% de las mujeres de 9 años seroconvirtieron a los VPH tipos 16 y 18, respectivamente, después de la tercer dosis (al mes 7) con TMG de, por lo menos, 1.4 a 2.4 veces más altos comparados con los de las mujeres de 10 a 14 años y 15 a 25 años, respectivamente. En dos estudios clínicos (HPV-012 y 013) realizados en niñas y adolescentes entre 10 y 14 años, todas las participantes seroconvirtieron a VPH tipos 16 y 18 después de la tercera dosis (al mes 7) con TMG de por lo menos 2 veces más altos que los de mujeres entre los 15 y 25 años. En un estudio clínico en curso (HPV-070) que se lleva a cabo en niñas de 9 a 14 años que reciben un programa de vacunación de 2 dosis (0, 6 meses ó 0, 12 meses), todas las participantes seroconvirtieron a ambos tipos de VPH 16 y 18 después de la segunda dosis. Se demostró que la respuesta inmune después de 2 dosis en mujeres de 9 a 14 años no era inferior a la respuesta inmune después de 3 dosis en mujeres de 15 a 25 años. La eficacia de Cervarix® se infiere sobre la base de los datos de inmunogenicidad observados en las niñas de 9 a 14 años de edad que fueron vacunadas. Inmunogenicidad en mujeres de 26 años o más: En el estudio de fase III (HPV-015) realizado en mujeres de 26 años o más, en el momento de evaluación a los 48 meses, es decir, 42 meses después de completar todo el ciclo de vacunación, el 100% y el 99.4% de las mujeres inicialmente seronegativas continuaron siendo seropositivas para los anticuerpos anti-VPH-16 y anti-VPH-18, respectivamente. Los títulos de anticuerpos tuvieron su valor máximo en el mes 7, disminuyeron gradualmente hasta el mes 18 y se estabilizaron hasta alcanzar una meseta al mes 48. En otro estudio clínico (HPV-014) realizado en mujeres de entre 26 y 55 años (N = 362), todas las participantes fueron seropositivas para los tipos de VPH 16 y 18 después de la tercera dosis (en el mes 7). Las TMG fueron menores en esta población, en comparación con las mujeres de 15 a 25 años. Sin embargo, todas las participantes permanecieron seropositivas para VPH-16 y todas, excepto una, permanecieron seropositivas para VPH- 18 a lo largo de la fase de seguimiento (hasta el mes 48) y mantuvieron niveles de anticuerpos en un orden de magnitud por encima de los que se encuentran después de la infección natural. Comparación de la inmunogenicidad de Cervarix® y Gardasil: En niñas de 9 a 14 años de edad: En un estudio comparativo con Gardasil (estudio HPV-071) realizado en niñas de 9 a 14 años, la superioridad de la respuesta inmune inducida por Cervarix® administrada según el esquema de vacunación de 2 dosis (0, 6 meses) comparado con el de Gardasil, que se administró según los esquemas de 2 dosis (0, 6 meses) y el estándar de 3 dosis (0, 2, 6 meses), quedó demostrado para VPH-16 y VPH-18 por ELISA (Tabla 6).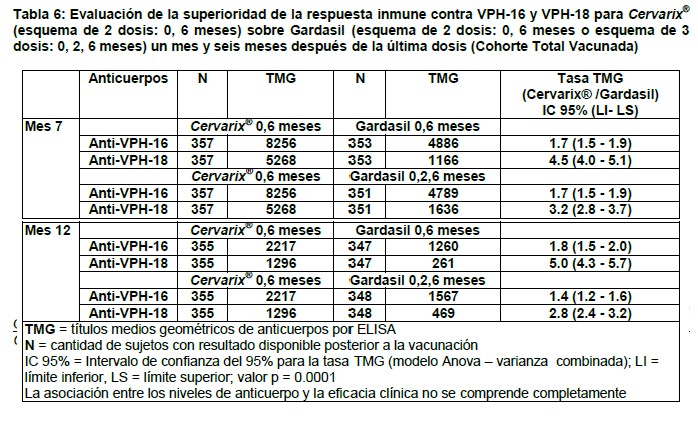
En mujeres de 18 a 45 años de edad: En un estudio comparativo de no inferioridad con Gardasil (estudio HPV-010) en mujeres de 18-45 años se demostró la no inferioridad de la respuesta inmune inducida por Cervarix® para los anticuerpos neutralizantes contra VPH-16 y VPH-18 en todas las cohortes etarias hasta tres años después de la primera vacunación (Tabla 7).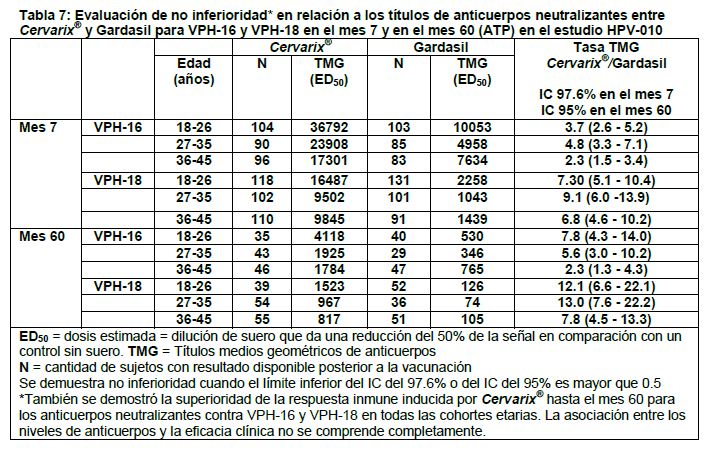
Inmunogenicidad en mujeres infectadas por el VIH: En un estudio clínico realizado en 120 sujetos asintomáticos con VIH positivo de 18 a 25 años de edad (60 sujetos recibieron Cervarix®), todas las participantes fueron seropositivas para VPH tipo 16 y para VPH tipo 18 después de la tercera dosis (en el mes 7), y la seropositividad para VPH tipos 16 y 18 se mantuvo hasta el mes 12. Los TMG son más bajos en esta población que los valores observados en los sujetos VIH negativos, pero la respuesta fue más de quince veces mayor que la respuesta a la infección natural por VPH, y los niveles de TMG fueron equivalentes o superiores a los niveles con los que se ha demostrado una eficacia sostenida. Se observó que, en general, Cervarix® es bien tolerado en las mujeres de 18 a 25 años infectadas por VIH durante un máximo de seis meses después de la última dosis de la vacuna y, durante el período de 12 meses del estudio, la vacuna no afectó el recuento de linfocitos CD4+, la carga viral de VIH ni el estadio clínico de la infección por VIH. Inmunogenicidad en varones de 10 a 18 años de edad: La inmunogenicidad en sujetos masculinos se evaluó en dos ensayos clínicos (estudio HPV-011 (N=173) y estudio HPV-040 (N=556). Los datos mostraron inmunogenicidad comparable en mujeres y varones. En el estudio HPV-011, todos los sujetos habían seroconvertido a VPH-16 y 18, y los niveles de TMG no fueron inferiores a los observados en los participantes femeninos de 15 a 25 años de edad del estudio HPV-012. Propiedades farmacocinéticas: No se requiere la evaluación de las propiedades farmacocinéticas de las vacunas. Datos de seguridad preclínica: Los datos no clínicos revelan la ausencia de peligros especiales para los seres humanos, basados en estudios convencionales de farmacología de seguridad, toxicidad con dosis agudas y repetidas, tolerabilidad local, fertilidad y toxicidad embrio-fetal y posnatal (hasta el final del período de lactancia).
Contraindicaciones: Cervarix® no deberá ser administrada a personas con hipersensibilidad conocida a cualquier componente de la vacuna (véanse Composición cualitativa y cuantitativa y Lista de excipientes).
Precauciones generales: Es buena práctica clínica que la vacunación sea precedida por una revisión de la historia médica (especialmente en cuanto a la vacunación previa y la ocurrencia posible de eventos adversos) y un examen clínico. Como con todas las vacunas inyectables, deberá contarse con el tratamiento médico y supervisión apropiados en caso de que ocurra una reacción anafiláctica, muy infrecuente, después de administrarse la vacuna. Puede presentarse síncope (desmayos) después, o incluso antes, de cualquier vacunación como una respuesta sicogénica a la inyección con aguja. Es importante que se tengan implementados los debidos procedimientos para evitar las lesiones por desmayos. Como ocurre con otras vacunas, la administración de Cervarix® deberá posponerse en las niñas y mujeres que padecen una afección febril grave y aguda. Sin embargo, la presencia de una infección menor, por ejemplo un resfriado, no debería ser motivo para aplazar la aplicación de la vacuna. En ningún caso deberá administrarse Cervarix® por vía intravascular o intradérmica. No se tienen datos sobre la administración subcutánea de Cervarix®. Al igual que en otras vacunas administradas por vía intramuscular, Cervarix® deberá administrarse con precaución a las personas con trombocitopenia o cualquier trastorno de la coagulación, puesto que en estos sujetos puede producirse una hemorragia después de la administración intramuscular. Como con cualquier vacuna, es posible que no se presente respuesta inmunoprotectora en todas las personas vacunadas. Cervarix® es una vacuna profiláctica. No busca prevenir el avance de las lesiones debidas al VPH presentes al momento de la vacunación. Cervarix® no ofrece protección frente a todos los tipos oncogénicos del VPH (ver propiedades farmacodinámicas). La vacunación es una prevención primaria y no es un substituto de los programas de detección temprana de anormalidades del cuello del útero (prevención secundaria), o de la adopción de precauciones contra la exposición al VPH y las enfermedades de transmisión sexual. Excepto por los sujetos con infección asintomática por el virus de inmunodeficiencia humana (VIH) de quienes se disponen datos limitados (ver Farmacodinamia), no hay información sobre el uso de Cervarix® en sujetos con respuesta inmune reducida, como los pacientes que reciben tratamiento inmunosupresor. Es posible que en estos pacientes no se obtenga la respuesta inmune adecuada. No se ha establecido completamente la duración de la protección. Se ha observado una eficacia protectora sostenida durante por lo menos 9.4 años después de la primera dosis. Se están realizando estudios a largo plazo para establecer la duración de la protección (ver propiedades farmacodinámicas). Efectos sobre la capacidad para conducir y operar maquinaria: No se han estudiado los efectos sobre la capacidad para conducir y utilizar máquinas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Se ha evaluado el efecto de Cervarix® sobre el desarrollo y sobrevivencia embrionario- fetal, perinatal y posnatal en ratas. Dichos estudios en animales no indican ningún efecto perjudicial directo o indirecto con respecto a la fertilidad, el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo posnatal. Los datos de mujeres embarazadas recopilados en ensayos clínicos, registros de embarazos y estudios epidemiológicos no sugieren que la vacunación con Cervarix® altera el riesgo de resultados anómalos en recién nacidos, tales como defectos de nacimiento. Los datos son insuficientes para concluir si la vacunación con Cervarix® afecta o no el riesgo de aborto espontáneo. Se recomienda a las mujeres embarazadas o que estén intentando quedar embarazadas posponer la vacunación hasta después del término del embarazo. Lactancia: El efecto en los lactantes cuyas madres recibieron Cervarix® durante el periodo de lactancia no se ha evaluado en estudios clínicos. Cervarix® podrá emplearse durante el período de lactancia únicamente cuando las posibles ventajas superen a los posibles riesgos. Los datos serológicos sugieren la transferencia de anticuerpos anti-VPH16 y anti-VPH18 a través de la leche durante el período de lactancia en ratas. Sin embargo, se desconoce si los anticuerpos inducidos por la vacuna se excretan en la leche materna humana.
Reacciones secundarias y adversas: Datos de estudios clínicos: En estudios clínicos, se administraron un total de aproximadamente 45,000 dosis de Cervarix® a aproximadamente 16,000 sujetos de 9-72 años, y se administraron aproximadamente 7,800 dosis a aproximadamente 2,600 sujetos masculinos de 10-18 años de edad. Se continuó estudiando a estos sujetos para evaluar la seguridad de la vacuna. La reacción observada con mayor frecuencia después de la administración de la vacuna fue dolor en el sitio de la inyección, que se produjo después del 78% de todas las dosis. La mayoría de estas reacciones tuvieron una intensidad leve o moderada, y no fueron de larga duración. Las reacciones adversas que se consideraron por lo menos posiblemente relacionadas con la vacunación se han clasificado por su frecuencia. Las frecuencias notificadas son las siguientes: Muy frecuentes (≥ 1/10). Frecuentes (≥ 1/100 a < 1/10) Infrecuentes (≥ 1/1,000 a < 1/100). Muy infrecuentes (≥ 1/10,000 a < 1/1,000). Infecciones e infestaciones: Infrecuentes: infección de las vías respiratorias altas. Trastornos del sistema sanguíneo y linfático: Infrecuente: linfadenopatía. Trastornos del sistema nervioso: Muy frecuentes: cefalea Infrecuentes: mareos. Trastornos digestivos: Frecuentes: gastrointestinales, incluidas las náuseas, vómitos, diarrea y dolor abdominal. Trastornos de la piel y de los tejidos blandos: Frecuentes: comezón/prurito, exantema, urticaria. Trastornos musculoesqueléticos y del tejido conjuntivo, y óseos: Muy frecuentes: mialgia. Frecuentes: artralgia. Trastornos generales y alteraciones en el lugar de administración: Muy frecuentes: reacciones en el sitio de la inyección, incluyendo dolor, rubor, tumefacción y fatiga. Frecuentes: fiebre (≥38°C). Infrecuentes: otras reacciones en el sitio de la inyección, como induración, parestesia local. Datos poscomercialización: Alteraciones en el sistema inmune: Muy infrecuentes: Reacciones alérgicas (incluyendo reacciones anafilácticas y anafilactoides), angioedema. Alteraciones en el sistema nervioso: Muy infrecuentes: Síncope o respuestas vasovagales a la inyección, algunas veces acompañados de movimientos tónico-clónicos.
Interacciones medicamentosas y de otro género: Uso con otras vacunas: Cervarix® puede administrarse de manera concomitante con cualquiera de las siguientes vacunas: vacuna antidiftérica-antitetánica-antipertussis acelular (dTpa) con contenido antigénico reducido, vacuna de poliovirus inactivados (IPV) y la vacuna combinada dTpa- IPV; vacuna de hepatitis A (inactivada) (HepA), vacuna de hepatitis B (rDNA) (HepB) y la vacuna HepA-HepB combinada. La administración de Cervarix® al mismo tiempo que Twinrix® (vacuna HepA-HepB combinada) no ha mostrado ninguna interferencia clínicamente relevante en la respuesta de anticuerpos frente a los antígenos VPH y de hepatitis A. Los títulos medios geométricos de los anticuerpos anti-HBs fueron más bajos en el caso de la coadministración, pero la importancia clínica de esta observación se desconoce debido a que los índices de seroprotección no se ven afectados. La proporción de sujetos que alcanzaron niveles de anticuerpos anti-HBs ≥10 mIU/ml fue de 98.3% para la vacunación concomitante y de 100% para Twinrix® solo. Si se va a administrar Cervarix® al mismo tiempo que otras vacunas inyectables, las vacunas deberán administrarse siempre en distintos sitios de inyección. Uso con anticonceptivos hormonales: En estudios de eficacia clínica, aproximadamente 60% de las mujeres tratadas con Cervarix® usaban anticonceptivos hormonales. No se tiene evidencia de que el uso de anticonceptivos hormonales afecte la eficacia de Cervarix®. Uso con medicamentos inmunosupresores sistémicos: Como ocurre con otras vacunas, puede esperarse que, en los pacientes que reciben tratamiento inmunosupresor, no se obtenga una respuesta adecuada. Incompatibilidades: A falta de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Alteraciones de los resultados de pruebas de laboratorio: No se han descrito hasta el momento.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los datos no clínicos revelan la ausencia de peligros especiales para los seres humanos, basados en estudios convencionales de farmacología de seguridad, toxicidad con dosis agudas y repetidas, tolerabilidad local, fertilidad y toxicidad embrio-fetal y posnatal (hasta el final del período de lactancia).
Dosis y vía de administración: Instrucciones para uso y manejo: Con el almacenamiento de la jeringa o vial, puede observarse un depósito blanco y fino, con un sobrenadante claro e incoloro. Esto no constituye un signo de deterioro. El contenido de la jeringa o vial deberá inspeccionarse visualmente tanto antes, como después de agitarse, para comprobar si hay presencia de partículas extrañas o un aspecto físico anormal antes de la administración. En caso de observar alguno de estos, elimínese la vacuna. La vacuna deberá agitarse bien antes de su uso. Vía de administración: Cervarix® se aplica como inyección intramuscular, en la región deltoidea (ver Precauciones generales e Interacciones medicamentosas y de otro género). Dosis: El esquema de vacunación dependerá de la edad del sujeto.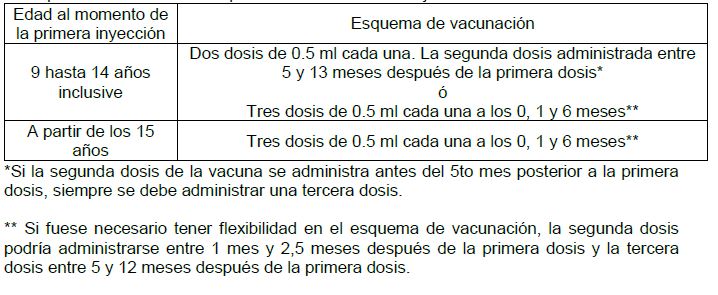
Aunque no se ha establecido la necesidad de una dosis de refuerzo, se ha observado que se produce una respuesta anamnésica después de la administración de una dosis adicional (ver Farmacodinamia). Instrucciones para la administración de la vacuna presentada en jeringa prellenada: 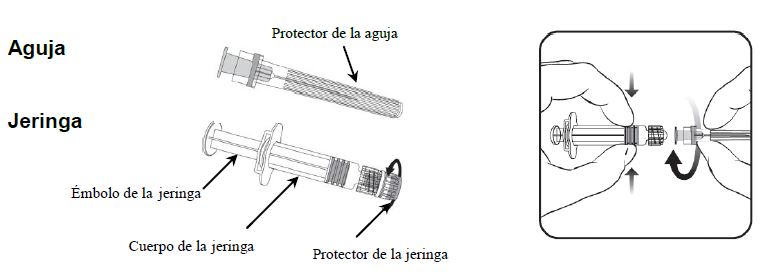
1. Sujetando el cuerpo de la jeringa con una mano (evite sujetar el émbolo de la jeringa), desenrosque el protector de la jeringa girándolo en sentido contrario de las manecillas del reloj. 2. Para acoplar la aguja a la jeringa, gire la aguja en el sentido de las manecillas del reloj en la jeringa hasta que la sienta bloqueada. (véase la ilustración). 3. Retire el protector de la aguja, que en ocasiones puede estar un poco apretado. 4. Administre la vacuna. Todo producto no utilizado o los materiales de desecho deberán eliminarse de acuerdo a los requerimientos locales. No todas las presentaciones están disponibles en todos los países.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Sobredosis: Los datos disponibles son insuficientes.
Presentaciones: Caja con 1,10 ó 100 frasco(s) ámpula con 1 dosis de 0.5 mL. Caja con 1 ó10 jeringa(s) prellenada(s) con 1 dosis de 0.5 mL. Naturaleza y contenido del envase: Cervarix® se presenta como una suspensión blanca turbia que al estar almacenada, se puede observar un depósito blanco y fino, con un sobrenadante claro e incoloro.
Recomendaciones sobre almacenamiento: Conservar en refrigeración (de +2 a +8 °C). NO CONGELAR. Conservar en el envase original a fin de proteger el producto de la luz. Cervarix® deberá administrarse tan pronto como sea posible una vez retirado del refrigerador. Sin embargo, los datos de estabilidad generados indican que la presentación de Cervarix® en envases monodosis se mantiene estable y puede administrarse, en caso de que se haya almacenado fuera del refrigerador, hasta tres días entre 8°C y 25°C ó hasta un día a temperaturas entre 25°C y 37°C.
Leyendas de protección: Literatura exclusiva para el Médico. Agítese antes de usarse. Deséchese la jeringa después de su uso. Léase instructivo anexo. No se administre este medicamento durante el embarazo y la lactancia. Su venta requiere receta médica. No se deje al alcance de los niños. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y farmacovigilancia.mx@gsk.com.
Nombre y domicilio del laboratorio: Hecho en Bélgica por: GlaxoSmithKline Biologicals S.A. Rue de l´Institut 89, 1330 Rixensart, Bélgica. Acondicionado y/o distribuido en México por: GlaxoSmithKline México, S.A. de C.V. Calz. México-Xochimilco No. 4900, Col. San Lorenzo Huipulco, C.P. 14370, México D.F.
Número de registro del medicamento: 369M2007 SSA IV.
Clave de IPP: No. de versión: GDS024/ IPI019 / EMISION: 2/MARZO/2015