EPAMIN®
PFIZER
Cápsulas
Denominación genérica: Fenitoína.
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: Fenitoína sódica 100 mg. Excipiente c.b.p. 1 Cápsula.
Indicaciones terapéuticas: Fenitoína está indicada para el control de crisis convulsivas tónico-clónicas generalizadas (gran mal) y de crisis parciales complejas (psicomotoras, lóbulo temporal), así como para la prevención y tratamiento de ataques ocurridos durante o posteriores a eventos neuroquirúrgicos. Fenitoína también ha sido usada en el tratamiento de la migraña, neuralgia trigeminal y ciertas psicosis. También ha sido usada en arritmias cardiacas, intoxicaciones con digitálicos y post-tratamiento en infarto al miocardio.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: La fenitoína es una droga anticonvulsivante útil en el tratamiento de la epilepsia. El sitio primario de acción parece ser la corteza motora donde se inhibe la diseminación de la actividad convulsivante. Posiblemente a través de la promoción del flujo de sodio de las neuronas, la fenitoína tiende a estabilizar el umbral contra la hiper-excitabilidad causada por la estimulación excesiva o los cambios ambientales capaces de reducir el gradiente de sodio de la membrana. Esto incluye la reducción de la potenciación post-tetánica a niveles sinápticos. La pérdida de la potenciación post-tetánica previene los focos de ataques corticales de las áreas corticales detonantes adyacentes. La fenitoína reduce la actividad máxima de los centros del tallo cerebral responsables de la fase tónica de los ataques tónicos-clónicos (gran mal). Propiedades farmacocinéticas: La fenitoína es un ácido débil y tiene una hidro-solubilidad limitada, aún en el intestino. El compuesto es absorbido en forma lenta y variable después de la administración oral. Una vez completada la absorción se distribuye rápidamente a todos los tejidos. La vida media plasmática promedio de fenitoína en el hombre es de 22 horas con un rango de 7 a 42 horas. Los niveles terapéuticos del equilibrio de la droga se alcanzan en 7 a 10 días después de iniciada la terapia con las dosis recomendadas de 300 mg/día. Con las formulaciones orales de fenitoína se obtienen niveles séricos máximos en 1½ - 3 horas después de la administración. La fenitoína tiene un volumen aparente de distribución de 0.6 L/kg con una elevada unión (90%) a las proteínas del plasma, principalmente a la albúmina. Los niveles de fenitoína libre pueden estar alterados en pacientes cuyas características de unión a proteínas difieren de lo normal. La fenitoína se distribuye al líquido cefalorraquídeo, saliva, semen, líquidos gástricos, bilis y leche materna. La concentración de fenitoína en el líquido cefalorraquídeo, cerebro y saliva se aproxima al nivel de la fenitoína libre en el plasma. La fenitoína es biotransformada en el hígado por el metabolismo oxidativo. La principal vía involucra la 4-hidroxilación, la cual representa 80% de todos los metabolitos, la enzima CYP 2C9 juega un papel importante en el metabolismo de la fenitoína (90% de la depuración intrínseca neta), mientras que CYP 2C19 tiene una participación menor en este proceso (10% de la depuración intrínseca neta). Esta contribución relativa de CYP 2C19 al metabolismo de fenitoína puede aumentar cuando las concentraciones de fenitoína son más altas. Debido a que el sistema del citocromo involucrado en la hidroxidación de la fenitoína en el hígado es saturable a concentraciones séricas elevadas, pequeños incrementos en la dosis de fenitoína pueden aumentar su vida media y producir cambios sustanciales en los niveles séricos cuando éstos se encuentran o exceden el rango terapéutico superior. El nivel en el equilibrio puede aumentar desproporcionadamente hasta dar como resultado una intoxicación cuando se incrementa la dosis en un 10% o más. La depuración de fenitoína se ve afectada por los inhibidores de CYP 2C9 tales como fenilbutazona y sulfametoxazol. También se ha observado un deterioro en la depuración en pacientes que han recibido inhibidores de CYP 2C19 como ticlopidina. La mayor parte de la droga es excretada en la bilis en forma de metabolitos inactivos que son luego reabsorbidos del tracto gastrointestinal y eliminados en la orina parcialmente a través de la filtración glomerular pero, aún más importante, por secreción tubular. Menos de 5% de la fenitoína es excretada en forma del compuesto parental. En la mayoría de los pacientes mantenidos con una dosis constante de una formulación oral, se han alcanzado niveles séricos estables de fenitoína. Existe una amplia variabilidad inter-paciente en los niveles séricos de fenitoína con dosis equivalentes. En los pacientes con niveles séricos inusualmente bajos puede deberse a falta de apego al tratamiento o a que son metabolizadotes ultrarrápidos de fenitoína. Generalmente, los niveles elevados son consecuencia de la enfermedad hepática, deficiencia enzimática congénita o interacciones con drogas que conducen a una interferencia metabólica. El paciente con amplias variaciones en los niveles séricos de fenitoína, a pesar de las dosis estándar, representa un problema clínico difícil. Las mediciones de los niveles séricos en dichos pacientes puede ser particularmente útiles. De ser necesarias, deben ser obtenidas 7-10 horas después de iniciado el tratamiento, después del cambio de la dosificación o de la adición o eliminación de otra droga al régimen para permitir lque se alcance un nuevo estado de equilibrio. Los niveles mínimos, obtenidos inmediatamente antes de la siguiente dosis programada del paciente, aportan información sobre el rango de niveles séricos clínicamente efectivos y confirman el cumplimiento terapéutico del paciente. Los niveles máximos de la droga, obtenidos en el momento de la concentración máxima esperada, indican el umbral individual de aparición de efectos colaterales relacionados con la dosis. Los estudios clínicos muestran que independientemente de si las tabletas son masticadas o no son bioequivalentes, producen niveles séricos aproximadamente similares y son absorbidas más rápidamente que las cápsulas de 100 mg. Interacción farmacocinética: La coadministración de tabletas de nelfinavir (1.250 mg dos veces al día) con la cápsula de fenitoína (200 mg una vez al día) no cambia la concentración plasmática de nelfinavir. Sin embargo, la coadministración de nelfinavir redujo los valores del ABC de fenitoína (total) y de fenitoína libre en 29% y 28% respectivamente.
Contraindicaciones: La fenitoína está contraindicada en pacientes hipersensibles a la fenitoína o a sus ingredientes inactivos o a otras hidantoínas, hipersensibilidad a componentes de acción efedrínica, pacientes coronarios graves, no está indicado en pacientes con pequeño mal epiléptico. No está indicado su uso durante la lactancia.
Precauciones generales: Advertencias Especiales y Precauciones Especiales para su Uso: General: La fenitoína no es efectiva en ataques de ausencia (pequeño mal). En presencia de ataques tónicos-clónicos (gran mal) y de ausencia (pequeño mal), se requiere la terapia combinada con otras drogas. La fenitoína no está indicada en convulsiones debidas a hipoglicemia o a otras causas metabólicas. Se requieren procedimientos de diagnóstico adecuados. La fenitoína no debe ser descontinuada en forma abrupta, debido a la posibilidad de aumento en la frecuencia de ataques, incluyendo estado de mal epiléptico. Si a criterio del médico, se requiere reducir la dosis, descontinuar o sustituir por una medicación antiepiléptica alterna, ésto debe hacerse gradualmente. Sin embargo, en el caso de reacción alérgica o de hipersensibilidad, puede ser necesaria la sustitución rápida por una terapia alterna. En este caso, la terapia alterna, debe ser una droga anticonvulsivante, no perteneciente a la clase química de las hidantoínas. Un pequeño porcentaje de individuos tratados con fenitoína, metabolizan la droga lentamente. El metabolismo lento, puede ser debido a una disponibilidad enzimática limitada y a la ausencia de inducción; lo cual parece estar determinado genéticamente (polimorfismo). La ingesta alcohólica aguda puede aumentar los niveles séricos de fenitoína, mientras la ingesta crónica de alcohol, puede reducir los niveles séricos. El Síndrome de Hipersensibilidad a Anticonvulsivantes (SHA), es un síndrome que aparece en raras ocasiones e inducido por fármacos, potencialmente fatal y ocurre en algunos pacientes que toman medicamentos anticonvulsivantes. Se caracteriza por fiebre, erupción dérmica (rash), linfoadenopatía y otras patologías multiorgánicas, a menudo hepáticas. El mecanismo es desconocido. El intervalo entre la primera exposición a la droga y la aparición de los síntomas es generalmente de 2 a 4 semanas, pero se ha reportado en individuos que han recibido anticonvulsivantes por 3 o más meses. Los pacientes con mayor riesgo de desarrollo de SHA, incluyen personas de raza negra, pacientes con historia familiar de aparición de este síndrome o quienes ya lo han experimentado con anterioridad, y pacientes inmunosuprimidos. El síndrome es más severo en individuos previamente sensibilizados. En pacientes con diagnóstico de SHA, debe de descontinuarse la fenitoína e iniciar las medidas de soporte apropiadas. Efecto sobre el Sistema Nervioso Central: Niveles séricos sostenidos de fenitoína, que excedan el rango óptimo, deben producir estados de confusión relacionados, tales como "delirio", "psicosis", o "encefalopatía" o una rara disfunción irreversible. En consecuencia, se recomienda la determinación de los niveles séricos de la droga, ante el primer signo de toxicidad aguda. Está indicada la reducción de la terapia con fenitoína si los niveles son excesivos; si persisten los síntomas, se recomienda concluir la terapia con fenitoína. Efecto hematopoyético: Algunos reportes han sugerido una relación entre la fenitoína y el desarrollo de linfoadenopatía (local o generalizada), incluyendo hiperplasia benigna de ganglios linfáticos, pseudolinfoma, linfoma y enfermedad de Hodgkin. Aunque no se ha establecido una relación causa y efecto, la presencia de linfoadenopatía indica la necesidad de diferenciar esta condición de otros tipos de patologías de los ganglios linfáticos. El compromiso de los ganglios linfáticos puede ocurrir, en presencia o no de signos y síntomas que se asemejan a la enfermedad del suero, es decir, fiebre, rash y compromiso hepático. En todos los casos de linfoadenopatía, se recomienda una observación de seguimiento por un período prolongado y realizar los esfuerzos adecuados para alcanzar el control de las convulsiones, con drogas anticonvulsivantes alternas. Aunque se ha observado macrocitosis y anemia megaloblástica, estas condiciones responden usualmente a la terapia con ácido fólico. Si se añade ácido fólico a la terapia con fenitoína, es posible que disminuya el control de las convulsiones. Efecto Hepático/Inmunológico: El hígado es el principal sitio de biotransformación de fenitoína. Los pacientes con deterioro de la función hepática, ancianos y pacientes gravemente enfermos, pueden mostrar signos de toxicidad. Se ha reportado hepatitis tóxica y daño hepático que pueden ser fatales en casos raros. Se han reportado casos de hepatotoxicidad aguda, incluyendo casos infrecuentes de insuficiencia hepática aguda con fenitoína. Estos incidentes se han asociado a un síndrome de hipersensibilidad, caracterizado por fiebre, erupciones cutáneas y linfoadenopatía, que usualmente ocurren durante los primeros dos meses de tratamiento. Otras manifestaciones comunes incluyen; artralgia, rash, ictericia, hepatomegalia, niveles séricos elevados de transaminasas, leucocitosis y eosinofilia. El curso clínico de la hepatotoxicidad aguda de la fenitoína, oscila desde la recuperación rápida a la evolución fatal. En los pacientes con hepatotoxicidad aguda, la fenitoína debe ser descontinuada inmediatamente y no readministrada. Algunos reportes de casos individuales han sugerido que es posible un aumento, pero aún raro, en la incidencia de reacciones de hipersensibilidad, incluyendo rash cutáneo y hepatotoxicidad, en pacientes de raza negra. Efecto Sobre el Sistema Tegumentario: La fenitoína puede causar en raras ocasiones eventos adversos cutáneos serios, tales como dermatitis exfoliativa, síndrome de Stevens-Johnson (SSJ) y necrolisis epidérmica tóxica (NET), que pueden ser fatales. Aunque pueden ocurrir reacciones cutáneas serias sin advertencias, los pacientes deben estar alertas sobre los signos y síntomas de rash cutáneo y aparición de vesículas, fiebre y otros signos de hipersensibilidad tales como prurito, y deben solicitar atención médica inmediatamente al observar cualquier signo o síntoma de este tipo. El médico debe advertir al paciente sobre la necesidad de descontinuar el tratamiento en presencia de cualquier erupción cutánea (rash). Si ésta es leve (tipo sarampión o escarlatiniforme), la terapia puede ser reiniciada una vez que el rash haya desaparecido completamente. Si el rash recurre al restituir la terapia, está contraindicado continuar el tratamiento con fenitoína. La literatura publicada ha sugerido la posibilidad de un aumento de riesgo de reacciones de hipersensibilidad incluyendo rash cutáneo, SSJ, NET, hepatotoxicidad y Síndrome de Hipersensibilidad a Anticonvulsivantes en pacientes de raza negra. Los estudios en pacientes descendientes de Chinos han mostrado una fuerte asociación entre el riesgo de desarrollo de SIS/NET y la presencia de HLA-B*1502, una variante alélica heredable del gen HLA B en pacientes que han utilizado otra carbamazepina. Evidencia limitada sugiere que el HLA-B*1502 puede ser un factor de riesgo del desarrollo de SIS/NET en pacientes con ascendencia asiática que tomen fármacos asociados a SIS/NET incluyendo fenitoína. Debe considerarse la posibilidad de evitar el uso de drogas asociadas a SIS/NET, incluyendo fenitoína, en pacientes HLA-B*1502 positivos cuando existan terapias alternas similares disponibles. Los reportes de la literatura sugieren que la combinación de fenitoína, irradiación craneal y la reducción gradual de los corticosteroides puede asociarse al desarrollo de eritema multiforme y/o síndrome de Stevens-Johnson y/o necrolisis epidérmica. Efecto Metabólico: En vista de los reportes aislados asociados a la fenitoína con exacerbación de la porfiria, se recomienda precaución en el uso de este medicamento en pacientes afectados de esta enfermedad. Se ha reportado hiperglicemia resultante de los efectos inhibidores de la droga sobre la liberación de insulina. La fenitoína también puede elevar los niveles séricos de glucosa en pacientes diabéticos. Efecto Musculoesquelético: Se piensa que la fenitoína y otros anticonvulsivantes que han mostrado inducción de la enzima CYP450 pueden afectar indirectamente el metabolismo mineral óseo incrementando el metabolismo de la vitamina D3. Esto puede conducir a una deficiencia de vitamina D y a un aumento en el riesgo de osteomalacia, fracturas óseas, osteoporosis, hipocalcemia y hipofosfatemia en pacientes epilépticos bajo tratamiento crónico. Información para el Paciente que usa la Formulación Oral de Fenitoína: Los pacientes que tomen fenitoína deben ser advertidos sobre la importancia en seguir en forma estricta el régimen de dosificación prescrito e informar a su médico sobre cualquier condición clínica en la que no sea posible tomar la droga prescrita por vía oral - por ejemplo, cirugía, etc. Los pacientes deben ser advertidos sobre el uso de un dispositivo de medición preciso al utilizar la formulación de suspensión oral. Los pacientes deben ser advertidos sobre el uso de otras drogas o bebidas alcohólicas sin informar previamente al médico. Los pacientes deben ser instruidos sobre la necesidad de llamar al médico en caso de que desarrollen rash cutáneo. Es importante la higiene dental adecuada con el fin de minimizar el desarrollo de hiperplasia de las encías y sus complicaciones.
Restricciones de uso durante el embarazo y la lactancia: Uso durante el embarazo: Algunos reportes sugieren una asociación entre el uso de drogas anticonvulsivantes por mujeres con epilepsia y una incidencia elevada de defectos del nacimiento en niños nacidos de estas madres. Los datos son más extensos en lo que respecta a la fenitoína y al fenobarbital, pero éstas son también las drogas anticonvulsivantes prescritas con mayor frecuencia. Reportes menos sistemáticos y anecdóticos sugieren una posible asociación similar con el uso de todas las drogas anticonvulsivantes conocidas. Los reportes que sugieren una mayor incidencia de defectos del nacimiento en niños de mujeres epilépticas tratadas con drogas no pueden ser considerados como adecuados para probar una relación causa y efecto definitiva. Existen problemas metodológicos intrínsecos en la obtención de datos adecuados sobre la teratogenicidad de las drogas en humanos. Los factores genéticos o la condición epiléptica misma pueden ser más importantes que la terapia con la droga en la etiología de los defectos del nacimiento. La gran mayoría de las madres bajo tratamiento anticonvulsivante dan a luz a niños normales. Es importante notar que las drogas anticonvulsivantes no deben ser descontinuadas en pacientes en quienes la droga es administrada para permitir convulsiones importantes debido a la fuerte posibilidad de precipitar un estado epiléptico con hipoxia y riesgo para la vida. Es posible considerar la descontinuación de la terapia antes y durante el embarazo en casos individuales en los que la severidad y la frecuencia de las convulsiones son tales que la suspensión de la medicación no expone al paciente a un riesgo serio, aunque no es posible establecer con confianza que aún un ataque menor no implique cierto riesgo para el embrión en desarrollo o el feto. El médico deberá ponderar estas consideraciones en el tratamiento y recomendaciones a la mujer epiléptica con potencial de procreación. Además de los reportes de una mayor incidencia malformaciones congénitas, tales como paladar/labio hendido y malformaciones cardíacas en niños de mujeres que reciben fenitoína y otras drogas anticonvulsivantes, también existen reportes de un síndrome de hidantoína fetal. Este consiste de una deficiencia del crecimiento prenatal, microencefalia y deficiencia fetal en niños nacidos de madres que han recibido fenitoína, barbitúricos, alcohol o trimetadiona. Sin embargo, estas características no están interrelacionadas y se asocian frecuentemente con un retardo del crecimiento intrauterino por otras causas. Se han registrado reportes aislados de procesos malignos, incluyendo neuroblastoma, en niños de madres que recibieron fenitoína durante el embarazo. En una alta proporción de pacientes la frecuencia de convulsiones aumenta durante el embarazo, debido a las alteraciones en la absorción y metabolismo de la fenitoína. La medición periódica de los niveles séricos de fenitoína es particularmente valiosa en el manejo de la paciente epiléptica embarazada como guía para el ajuste apropiado de la dosis. Sin embargo, probablemente esté indicada la restauración post-parto de la dosificación original. Se han reportado defectos neonatales en la coagulación durante las primeras 24 horas en niños nacidos de madres epilépticas que han recibido fenobarbital y/o fenitoína. La vitamina K ha mostrado prevenir o prevenir este defecto y ha sido recomendada su administración a la madre antes del alumbramiento y al neonato después del nacimiento. Uso en madres en lactancia: No se recomienda la lactancia materna del infante de madres que tomen esta droga debido a que la fenitoína parece ser secretada a bajas concentraciones en la leche humana. La concentración de fenitoína en una leche materna es aproximadamente un tercio de la concentración correspondiente en el plasma materno.
Reacciones secundarias y adversas: Efectos indeseables: Organismo como un todo: Reacción anafilactoide y anafilaxis. Sistema nervioso central: Las manifestaciones más comunes encontradas durante la terapia con fenitoína son debidas a este sistema y usualmente se relaciona con la dosis. Estas incluyen nistagmus, ataxia, lenguaje confuso, disminución de la coordinación y confusión mental. (véase Advertencias especiales y Precauciones especiales de uso - Efectos sobre el sistema nervioso central). También se ha reportado vértigo, insomnio, nerviosismo transitorio, espasmo motor, cefalea, parestesia, y somnolencia. También existen reportes de discinesia inducida por la fenitoína, incluyendo corea, distonía, temblor y asterixis, similar a las inducidas por la fenotiazina y otras drogas neurolépticas. Se ha observado raras veces una polineuropatía periférica predominantemente sensorial en pacientes que han recibido terapia a largo plazo con fenitoína. Sistema del tejido conectivo: Engrosamiento de las características faciales, engrosamiento de los labios, hiperplasia gingival, hipertricosis y enfermedad de Peyronie. Sistema gastrointestinal: Náusea, vómito, constipación, hepatitis tóxica y daño hepático (véase Advertencias especiales y Precauciones especiales de uso - Efecto hepático/inmunológico). Sistema hematopoyético: Se han reportado ocasionalmente complicaciones hematopoyéticas, algunas veces fatales, en asociación con la administración de fenitoína. Estas han incluido trombocitopenia, leucopenia, granulocitopenia, agranulocitosis y pancitopenia con supresión o no en la médula ósea. También se han reportado macrocitosis y anemia megaloblástica. Igualmente se ha observado linfoadenopatía, incluyendo hiperplasia benigna de ganglios linfáticos, seudolinfoma, linfoma y enfermedad de Hodgkin. (Véase Advertencias especiales y Precauciones especiales de uso - Efecto hematopoyético). Inmunológico: Síndrome de hipersensibilidad, lupus eritematoso sistémico, periarteritis nodosa y anormalidades en las inmunoglobulinas (véase Sección Advertencias especiales y Precauciones especiales de uso - Efecto hepático/inmunológico). Sistema tegumentario: Las manifestaciones dermatológicas, algunas veces acompañadas de fiebre han incluido rash escarlatiniforme o morbiliforme. El rash morbiliforme (tipo sarampión) es el más común; otros tipos de dermatitis son más raros. Otras formas más serias que pueden ser fatales incluyen dermatitis bulosa, exfoliativa o purpúrea, lupus eritematoso, síndrome de Stevens-Johnson y necrolisis epidérmica. (véase Sección Advertencias especiales y Precauciones especiales de uso - Efecto tegumentario). Sentidos especiales: Perversión del gusto. Experiencia post-mercadeo: Sistema musculoesquelético: Se han asociado fracturas óseas y osteomalacia con el uso a largo plazo ( > 10 años) de fenitoína en pacientes con epilepsia crónica. También se ha reportado osteoporosis y otros trastornos del metabolismo óseo tales como hipocalcemia, hipofosfatemia y reducción de los niveles de metabolitos de vitamina D.
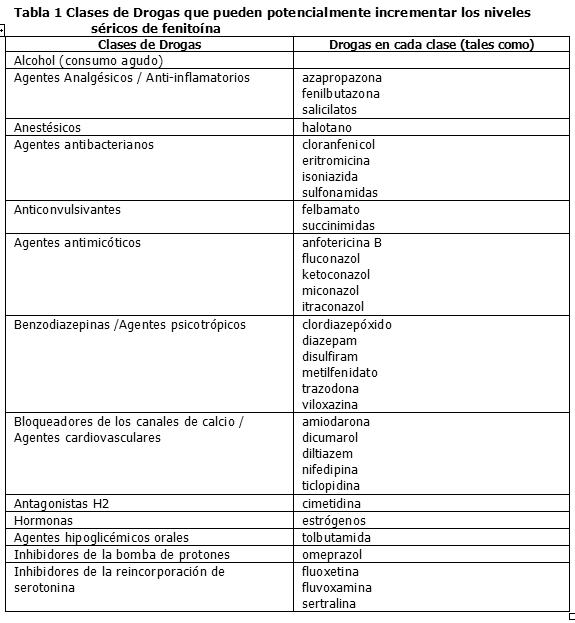
Interacciones medicamentosas y de otro género: Interacción con otros productos medicinales y otras formas de interacción: Interacciones entre drogas: Muchas drogas pueden aumentar o disminuir los niveles séricos de fenitoína o sus efectos. Las determinaciones de las concentraciones séricas de fenitoína son particularmente útiles si se sospecha de posibles interacciones con drogas. Las interacciones entre drogas más comunes se indican a continuación. Drogas que pueden incrementar los niveles séricos de fenitoína: Varias drogas pueden aumentar los niveles séricos de fenitoína o disminuir su tasa de metabolismo por los sistemas enzimáticos hepáticos CYP450 2C9 y 2C19 (por ejemplo, dicumarol, disulfiram, omeprazol, ticlopidina), por competencia por los sitios de unión a las proteínas (por ejemplo salicilatos, sulfisoxazol, tolbutamida) o por una combinación de ambos procesos (por ejemplo fenilbutazona, valproato sódico) (véase Tabla 1).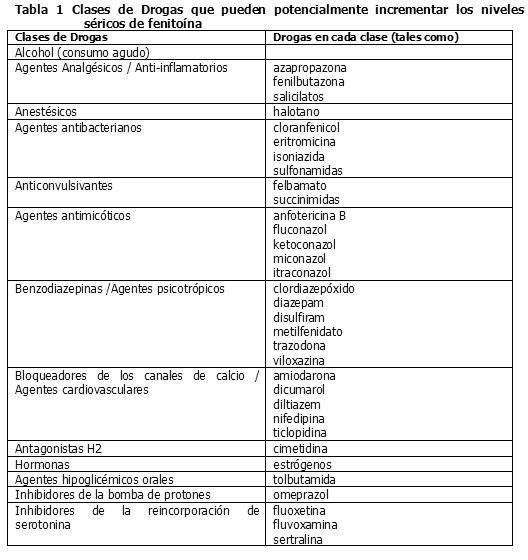
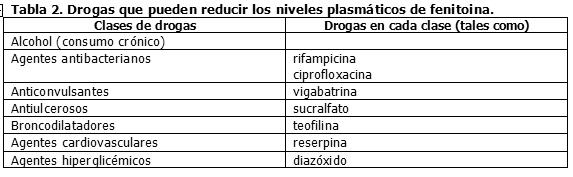
Drogas que pueden reducir los niveles séricos de fenitoína: La Tabla 2 resume las clases de drogas que pueden potencialmente disminuir los niveles plasmáticos de fenitoína.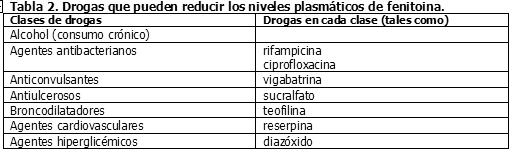
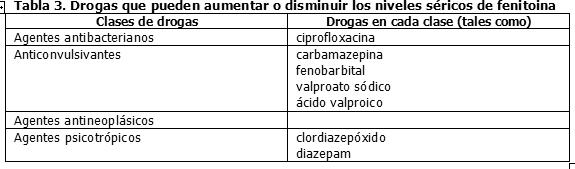
El clorhidrato de molindona contiene iones de calcio que interfieren con la absorción de fenitoína. Los tiempos de ingestión de fenitoína y de preparaciones de calcio, incluyendo preparaciones antiácidas que contengan calcio, deben ser distanciados para prevenir los problemas de absorción. Un estudio de interacción farmacocinética entre nelfinavir y fenitoína, ambos administrados oralmente, mostró que nelfinavir redujo los valores de ABC de la fenitoína (total) y fenitoína libre en 29% y 28% respectivamente. En consecuencia, la concentración de fenitoína debe ser monitoreada durante la coadministración de nelfinavir, ya que nelfinavir puede reducir la concentración plasmática de fenitoína (véase Sección Propiedades Farmacocinéticas - Interacción Farmacocinética). Drogas que pueden aumentar o disminuir los niveles séricos de fenitoína: La Tabla 3 resume las clases de drogas que pueden aumentar o disminuir los niveles séricos de fenitoína.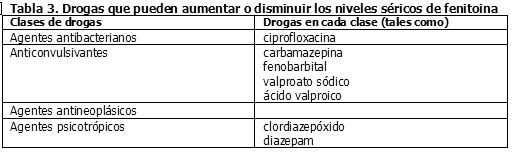
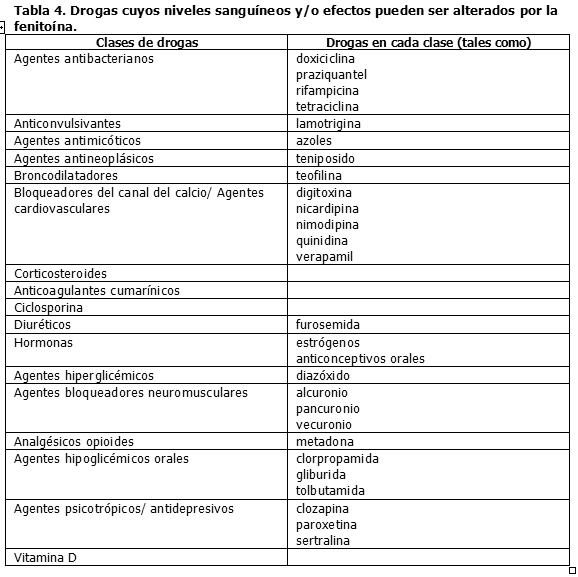
Similarmente, los efectos de la fenitoína sobre los niveles séricos de carbamazepina, fenobarbital, ácido valproico y valproato sódico, es impredecible. Drogas cuyos niveles sanguíneos y/o efectos pueden ser alterados por la fenitoína. La Tabla 4 resume las clases de drogas cuyos niveles sanguíneos y/o efectos pueden alterados por la fenitoína: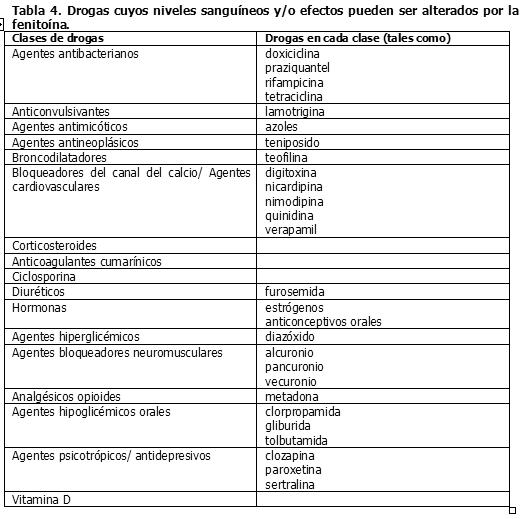
Aunque no es realmente una interacción entre drogas, los antidepresivos tricíclicos pueden precipitar las convulsiones en pacientes susceptibles por lo que puede ser necesario el ajuste de la dosificación de fenitoína. Interacción entre fármacos y alimentación parenteral/preparaciones nutricionales: Los reportes de la literatura sugieren que los pacientes que han recibido preparaciones de alimentación parenteral y/o suplementos nutricionales relacionados tienen niveles plasmáticos de fenitoína más bajos que los esperados. En consecuencia se sugiere que la fenitoína no sea administrada concomitantemente con preparaciones de alimentación parenteral. Puede ser necesario el monitoreo más frecuente de los niveles séricos de fenitoína en estos pacientes.
Alteraciones en los resultados de pruebas de laboratorio: La fenitoína puede causar una reducción en los niveles séricos de yodo unido a proteínas (PBI). También puede producir valores menores que los normales en las pruebas de dexametasona o metirapona. La fenitoína puede causar aumento en los niveles séricos de glucosa, fosfatasa alcalina y gamma glutamil transpeptidasa (GGT). La fenitoína puede afectar los niveles sanguíneos de calcio y las tasas de metabolismo de azúcar en sangre.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Véase Precauciones o restricciones de uso durante el embarazo y la lactancia. Efectos sobre la capacidad de conducir y operar maquinarias: Los pacientes deben ser advertidos de que no deben conducir automóviles u operar maquinaria potencialmente peligrosa hasta que se establezca que este medicamento no afecta su capacidad de realizar estas actividades.
Dosis y vía de administración: Vía de Administración: Oral. Posología y método de administración: Generalidades: Las cápsulas y la solución para inyección de fenitoína, están formuladas con la sal sódica de fenitoína. La forma ácida libre de la fenitoína es utilizada en las forma de suspensión (30 mg/5 mL (pediátrico) y 125 mg/5 mL) y en las tabletas de fenitoína. Debido a un aumento de aproximadamente 8% en el contenido de la droga de la forma ácida libre, en comparación con la sal sódica, pueden ser necesarios ajustes de la dosis y el monitoreo del nivel sérico al cambiar desde un producto formulado con la sal libre a un producto formulado con la sal sódica y viceversa. Para todas las formulaciones orales, la dosis debe ser individualizada para obtener el beneficio máximo. En algunos casos puede ser necesaria la determinación de los niveles séricos de la droga para realizar ajustes de dosificación óptimos. El control adecuado sin signos clínicos de toxicidad, es más frecuente con niveles séricos comprendidos entre 10-20 mcg/mL, aunque los casos leves epilepsia tónica-clónica (gran mal) pueden ser controlados con niveles séricos más bajos de fenitoína. Con la dosificación recomendada, puede ser necesario un período de 7 a 10 días para alcanzar niveles séricos en el equilibrio con fenitoína y los cambios en la dosis (incremento o reducción) no deben ser realizados a intervalos menores de 7 a 10 días. Dosificación en Adultos: Dosis diaria dividida: En el caso de las tabletas o cápsulas orales, los pacientes que no hayan recibido tratamiento previo, pueden iniciar con una dosis de 300 mg al día, a ser tomada en 3 dosis igualmente divididas y la dosificación puede ser luego ajustada a los requerimientos individuales. Para la mayoría de los adultos, la dosis satisfactoria de mantenimiento será de 300 mg ó 400 mg al día, a ser tomadas en e 3 ó 4 dosis igualmente divididas respectivamente. De ser necesario, es posible aumentar hasta 600 mg al día. Dosis oral de carga (impregnación) en pacientes adultos en ausencia de una emergencia. Es aquellos adultos que requieran, en ausencia de una emergencia, alcanzar rápidamente los niveles séricos en estado estacionario, o en quienes no es deseable la administración intravenosa, se puede utilizar una dosis de carga de fenitoína. Este régimen de dosificación debe ser reservado para pacientes hospitalizados, en quienes es posible monitorear los niveles séricos de fenitoína. Los pacientes con historia de enfermedad renal o hepática, no deben recibir un régimen de dosis de carga oral. La dosis de carga oral recomendada es de 1 gramo de fenitoína, dividida en 3 dosis (400 mg, 300 mg, 300 mg) administradas con intervalos de dos horas. Veinticuatro horas después de la dosis de carga se instituye la dosis de mantenimiento normal con determinaciones frecuentes de los niveles séricos. Dosificación pediátrica: En el caso de las tabletas o cápsulas orales, inicialmente 5 mg/kg/día en 2 ó 3 dosis iguales divididas, con la individualización subsiguiente de la dosis hasta un máximo de 300 mg al día. La dosificación diaria de mantenimiento recomendada, es usualmente de 4 a 8 mg/kg. Niños mayores de 6 años y adolescentes, pueden requerir la dosis mínima del adulto (300 mg/día). Si no es posible dividir la dosis diaria en cantidades iguales, la mayor dosis debe ser administrada al acostarse.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Sobredosis: Se desconoce la dosis letal en pacientes pediátricos. La dosis letal en adultos se ha estimado en 2 a 5 gramos. Los síntomas iniciales son nistagmus, ataxia y disartria. Otros signos son temblores, hiperreflexia, somnolencia, letargia, lenguaje confuso, visión borrosa, náusea y vómito. El paciente puede desarrollar coma e hipotensión. La muerte se debe a la depresión respiratoria y circulatoria. Existen amplias variaciones entre los individuos con respecto a los niveles séricos de fenitoína en los que puede ocurrir toxicidad. El nistagmus en posición decúbito se observa usualmente a 20 mcg/ml y ataxia a 30 mcg/ml. La disartria y la letargia ocurren a concentraciones séricas > 40 mcg/ml, pero se han reportado concentraciones tan elevadas como de 50 mcg/ml sin evidencias de toxicidad. Se han ingerido tanto como 25 veces la dosis terapéutica que produjeron concentraciones séricas > 100 mcg/ml con recuperación completa. Tratamiento: El tratamiento no es específico ya que no existe un antídoto conocido. Es necesario observar cuidadosamente la función respiratoria y circulatoria y emplear las medidas de soporte apropiadas. Es posible considerar la hemodiálisis ya que la fenitoína no se une completamente a las proteínas del plasma. Se han utilizado transfusiones totales de sangre en el tratamiento de la intoxicación severa en pacientes pediátricos. En la sobredosis aguda, debe tomarse en cuenta la posibilidad de la presencia de otros depresores del SNC, incluyendo alcohol.
Presentación(es): Caja con 50 cápsulas de 100 mg en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. El empleo de este medicamento durante el embarazo, queda bajo la responsabilidad del médico.
Nombre y domicilio del laboratorio: Pfizer, S.A. de C.V. Km 63. Carretera México-Toluca, Zona Industrial, C.P. 50140, Toluca, México, México. ®Marca Registrada
Número de registro del medicamento: 21502 SSA IV
Clave de IPPA: 093300415D0272
EPAMIN®
PFIZER
Suspensión
Denominación genérica: Fenitoína.
Forma farmacéutica y formulación: Suspensión. Cada 100 ml contiene: Fenitoína 0.7500 g y 2.5 g. Excipiente c.b.p. 100.0 mL. (Una cucharadita de 5 ml contiene el equivalente a 37.5 mg ó 125 mg de fenitoina).
Indicaciones terapéuticas: La fenitoína está indicada para el control de las crisis generalizadas tónico-clónicas (gran mal) y convulsiones parciales complejas (psicomotoras, del lóbulo temporal), y en la prevención y tratamiento de convulsiones que aparecen durante o después de neurocirugía.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: La fenitoína es una droga anticonvulsivante útil en el tratamiento de la epilepsia. El sitio primario de acción parece ser la corteza motora donde se inhibe la diseminación de la actividad convulsivante. Posiblemente a través de la promoción del flujo de sodio de las neuronas, la fenitoína tiende a estabilizar el umbral contra la hiper-excitabilidad causada por la estimulación excesiva o los cambios ambientales capaces de reducir el gradiente de sodio de la membrana. Esto incluye la reducción de la potenciación post-tetánica a niveles sinápticos. La pérdida de la potenciación post-tetánica previene los focos de ataques corticales de las áreas corticales detonantes adyacentes. La fenitoína reduce la actividad máxima de los centros del tallo cerebral responsables de la fase tónica de los ataques tónicos-clónicos (gran mal). Propiedades farmacocinéticas: La fenitoína es un ácido débil y tiene una hidro-solubilidad limitada, aún en el intestino. El compuesto es absorbido en forma lenta y variable después de la administración oral. Una vez completada la absorción se distribuye rápidamente a todos los tejidos. La vida media plasmática promedio de fenitoína en el hombre es de 22 horas con un rango de 7 a 42 horas. Los niveles terapéuticos del equilibrio de la droga se alcanzan en 7 a 10 días después de iniciada la terapia con las dosis recomendadas de 300 mg/día. Con las formulaciones orales de fenitoína se obtienen niveles séricos máximos en 1½ - 3 horas después de la administración. La fenitoína tiene un volumen aparente de distribución de 0.6 L/kg con una elevada unión (90%) a las proteínas del plasma, principalmente a la albúmina. Los niveles de fenitoína libre pueden estar alterados en pacientes cuyas características de unión a proteínas difieren de lo normal. La fenitoína se distribuye al líquido cefalorraquídeo, saliva, semen, líquidos gástricos, bilis y leche materna. La concentración de fenitoína en el líquido cefalorraquídeo, cerebro y saliva se aproxima al nivel de la fenitoína libre en el plasma. La fenitoína es biotransformada en el hígado por el metabolismo oxidativo. La principal vía involucra la 4-hidroxilación, la cual representa 80% de todos los metabolitos, la enzima CYP 2C9 juega un papel importante en el metabolismo de la fenitoína (90% de la depuración intrínseca neta), mientras que CYP 2C19 tiene una participación menor en este proceso (10% de la depuración intrínseca neta). Esta contribución relativa de CYP 2C19 al metabolismo de fenitoína puede aumentar cuando las concentraciones de fenitoína son más altas. Debido a que el sistema del citocromo involucrado en la hidroxidación de la fenitoína en el hígado es saturable a concentraciones séricas elevadas, pequeños incrementos en la dosis de fenitoína pueden aumentar su vida media y producir cambios sustanciales en los niveles séricos cuando éstos se encuentran o exceden el rango terapéutico superior. El nivel en el equilibrio puede aumentar desproporcionadamente hasta dar como resultado una intoxicación cuando se incrementa la dosis en un 10% o más. La depuración de fenitoína se ve afectada por los inhibidores de CYP 2C9 tales como fenilbutazona y sulfametoxazol. También se ha observado un deterioro en la depuración en pacientes que han recibido inhibidores de CYP 2C19 como ticlopidina. La mayor parte de la droga es excretada en la bilis en forma de metabolitos inactivos que son luego reabsorbidos del tracto gastrointestinal y eliminados en la orina parcialmente a través de la filtración glomerular pero, aún más importante, por secreción tubular. Menos de 5% de la fenitoína es excretada en forma del compuesto parental. En la mayoría de los pacientes mantenidos con una dosis constante de una formulación oral, se han alcanzado niveles séricos estables de fenitoína. Existe una amplia variabilidad inter-paciente en los niveles séricos de fenitoína con dosis equivalentes. En los pacientes con niveles séricos inusualmente bajos puede deberse a falta de apego al tratamiento o a que son metabolizadotes ultrarrápidos de fenitoína. Generalmente, los niveles elevados son consecuencia de la enfermedad hepática, deficiencia enzimática congénita o interacciones con drogas que conducen a una interferencia metabólica. El paciente con amplias variaciones en los niveles séricos de fenitoína, a pesar de las dosis estándar, representa un problema clínico difícil. Las mediciones de los niveles séricos en dichos pacientes puede ser particularmente útiles. De ser necesarias, deben ser obtenidas 7-10 horas después de iniciado el tratamiento, después del cambio de la dosificación o de la adición o eliminación de otra droga al régimen para permitir que se alcance un nuevo estado de equilibrio. Los niveles mínimos, obtenidos inmediatamente antes de la siguiente dosis programada del paciente, aportan información sobre el rango de niveles séricos clínicamente efectivos y confirman el cumplimiento terapéutico del paciente. Los niveles máximos de la droga, obtenidos en el momento de la concentración máxima esperada, indican el umbral individual de aparición de efectos colaterales relacionados con la dosis. Los estudios clínicos muestran que independientemente de si las tabletas son masticadas o no son bioequivalentes, producen niveles séricos aproximadamente similares y son absorbidas más rápidamente que las cápsulas de 100 mg. Interacción farmacocinética: La coadministración de tabletas de nelfinavir (1.250 mg dos veces al día) con la cápsula de fenitoína (200 mg una vez al día) no cambia la concentración plasmática de nelfinavir. Sin embargo, la coadministración de nelfinavir redujo los valores del ABC de fenitoína (total) y de fenitoína libre en 29% y 28% respectivamente.
Contraindicaciones: La fenitoína está contraindicada en pacientes hipersensibles a la fenitoína, o a los ingredientes inactivos del producto, o a otras hidantoínas.
Precauciones generales: Advertencias Especiales y Precauciones Especiales para su U