ETONIRI
SANFER
Denominación genérica: Irinotecan.
Forma farmacéutica y formulación: Cada frasco ámpula de solución contiene: Clorhidrato de irinotecan 100 mg Vehículo, c.b.p. 5 ml
Indicaciones terapéuticas: ETONIRI® (irinotecan) está indicado como agente único o combinado en el tratamiento de pacientes con: cáncer de colon y recto metastásico recurrente o progresivo, cáncer gástrico, cáncer de ovario, cáncer de pulmón en células pequeñas y no pequeñas, cáncer cervicouterino, cáncer de esófago, cáncer gástrico inoperable o recurrente. ETONIRI® (irinotecan) está indicado como agente único en Eel tratamiento de pacientes con: cáncer de mama metastásico o recurrente, carcinoma cutáneo de células escamosas, melanoma maligno, cáncer de páncreas, glioma.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: Clase terapéutica: ETONIRI® (Clorhidrato de irinotecan) es un agente antineoplásico del grupo de los inhibidores de la topoisomerasa I, clínicamente investigado como CPT-11. El irinotecan es un derivado semisintético de la camptotecina, un alcaloide extraído de plantas como Camptotheca acuminata. Mecanismo de acción: El irinotecan y su metabolito activo SN-38 se unen al complejo de la ADN-topoisomerasa I y previenen la religación de las hebras simples divididas de la banda simple. Las investigaciones actuales sugieren que la citotoxicidad del irinotecan es debida al daño que produce en la doble hebra del ADN durante la síntesis, cuando las enzimas de replicación interactúan con el complejo ternario formado por la topoisomerasa I, ADN e irinotecan o SN-38, cualquiera de los dos. El irinotecan sirve como un precursor soluble en agua del metabolito lipofílico SN-38. El SN-38 es formado a partir del irinotecan por la carboxiesterasa, mediante la separación de la unión entre la porción de camptotecina y la cadena lateral de dipiperidino. El SN-38 es aproximadamente 1,000 veces más potente que el irinotecan como un inhibidor de la topoisomerasa I purificado de líneas celulares de tumores de humanos y roedores. Los ensayos de citotoxicidad in vitro muestran que la potencia del SN-38 relacionada con el irinotecan varía de 2 a 2,000 veces; el SN-38 se une aproximadamente en 95% a las proteínas plasmáticas comparado con irinotecan que se une aproximadamente en 50%, a las proteínas plasmáticas. La contribución precisa del SN-38 al irinotecan es todavía desconocida. Ambos existen en forma de lactona activa y de un anión hidroxiácido. Un equilibrio dependiente del pH existe entre las dos formas, de manera que el pH ácido promueve la formación de lactona, mientras que un pH más básico favorece la formación de un anión hidroxiácido. Propiedades farmacocinéticas: Absorción y distribución: Después de la infusión intravenosa en humanos, las concentraciones en plasma del irinotecan declinan en una manera exponencial, con una vida media de eliminación terminal de aproximadamente 6 horas. La vida media de eliminación terminal del metabolito activo SN-38 es de aproximadamente 10 horas. Las vidas medias de las formas de lactona activa del irinotecan y del SN-38 son similares a las de los compuestos primarios, las formas de lactona y de hidroxiácido se encuentran en equilibrio. Sobre el intervalo de dosificación de 50 a 350 mg/m², el área bajo la curva (ABC) de irinotecan se incrementa linealmente con la dosis; el ABC del SN-38 se incrementa menos que proporcionalmente con la dosis. Las concentraciones máximas del metabolito activo SN-38 son generalmente observadas después de una hora posterior al final de una infusión de 90 minutos de irinotecan. El irinotecan muestra una unión moderada a las proteínas del plasma (30-68%); mientras que el SN-38 se une altamente principalmente a la albúmina.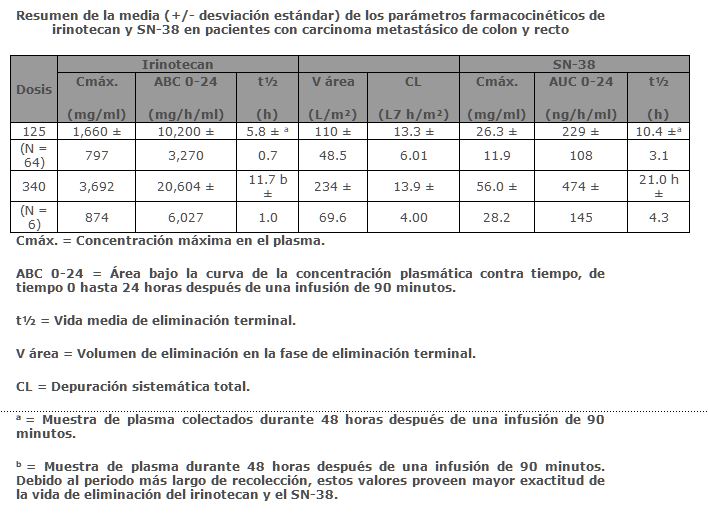
Metabolismo y excreción: La conversión metabólica del irinotecan al metabolito activo SN-38 está mediada por enzimas carboxilesterasas y ocurre primariamente en el hígado. El SN-38 subsecuentemente sufre una conjugación para formar un metabolito glucurónido.El SN-38 glucurónido tiene 1/50 a 1/100 de actividad del SN-38 en ensayos de citotoxicidad utilizando líneas celulares para estudios in vitro. La disposición del irinotecan no ha sido completamente medida en humanos. La excreción urinaria del irinotecan es de 11% a 20%; SN-38, < 1%; y SN-38 glucurónido 3% respectivamente. La excreción acumulativa biliar y urinaria de irinotecan y sus metabolitos sobre un periodo de 48 horas después de la administración de irinotecan en dos pacientes, se encuentra en el intervalo de aproximadamente 25% (100 mg/m²) a 50% (300 mg/m²).Farmacocinéticas en grupos de edad:Geriátricos: La vida media terminal del irinotecan fue de 6.0 horas en pacientes de 65 o más años y de 5.5 horas en pacientes más jóvenes de 65 años. El ABC O-24 para SN-38 en pacientes que tenían al menos 65 años de edad fue 11% más alta que en pacientes menores de 65 años. No se recomiendan cambios en la dosis y administración para pacientes geriátricos.Pediátricos: La farmacocinética de irinotecan no ha sido estudiada en poblaciones pediátricas.Género: La farmacocinética del irinotecan no parece estar influenciada por el género.Raza: La influencia de la raza en la farmacocinética del irinotecan no ha sido evaluada.Insuficiencia hepática: (Véase Dosis y vía de administración, poblaciones especiales). La depuración de irinotecan es reducida en pacientes con disfunción hepática con un aumento relativo a la exposición del metabolito activo SN-38. La magnitud de esos efectos es proporcional al grado del deterioro del hígado medido por el aumento en suero de la concentración de bilirrubina total y transaminasas.Insuficiencia renal: La influencia de la insuficiencia renal en la farmacocinética del irinotecan no ha sido evaluada (véase Dosis y vía de administración, Pacientes con función renal deteriorada).
Contraindicaciones: ETONIRI® irinotecan está contraindicado en pacientes con hipersensibilidad conocida a la droga o a sus excipientes (véase Precauciones generales y reacciones de hipersensibilidad).
Precauciones generales: Administración: El irinotecan solamente se debe administar por vía intravenosa y bajo la supervisión de un médico con experiencia en la administración de agentes quimioterapéuticos y especialista en oncología. Solamente es posible el manejo adecuado de las complicaciones cuando se dispone de equipo e instalaciones para el tratamiento.Irinotecan sólo será prescrito en los siguientes casos después de que los beneficios esperados se hayan analizado contra los posibles riesgos terapéuticos.En pacientes que presenten un factor de riesgo, particularmente aquellos con un estado de funcionamiento ECOG = 2 o menor. En unas cuantas y raras instancias donde se estima como poco probable que los pacientes observen las recomendaciones con respecto al manejo de eventos adversos (la necesidad del tratamiento antidiarreico inmediato y prolongado combinado con la ingesta alta de fluidos en el inicio de la diarrea tardía). Se recomienda una estricta supervisión hospitalaria para estos pacientes.Síntomas colinérgicos: Los pacientes pueden tener síntomas colinérgicos de rinitis, salivación aumentada, miosis, lagrimeo, diaforesis, enrojecimiento (vasodilatación), bradicardia e hiperperistaltismo intestinal que puede causar calambres abdominales y diarrea temprana (es decir, la diarrea que generalmente ocurre durante las primeras 8 horas a la administración del irinotecan). Se cree que estos síntomas, los cuales se pueden observar durante o poco después de la infusión del irinotecan, están relacionados con la actividad anticolinérgica del irinotecan inalterado, y se espera que ocurran más frecuentemente con dosis mayores de irinotecan.En los pacientes con síntomas colinérgicos se debe considerar la administración terapéutica o profiláctica de 0.25 a 1 mg de atropina intravenosa o subcutánea (a menos que esté contraindicada). Extravasación: Aunque el irinotecan no es un vesicante conocido, se debe tener cuidado de evitarla. Si ocurre ésta; se recomienda lavar el sitio con abundante agua y jabón y aplicar hielo.Reacciones de hipersensibilidad: Se han reportado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas/anafilactoides severas.Diarrea: El irinotecan puede inducir tanto diarrea temprana como tardía, las cuales al parecer están mediadas por diferentes mecanismos, y ambas pueden ser severas. La diarrea temprana de naturaleza colinérgica (se presenta durante o poco después de la administración) puede estar acompañada de síntomas colinérgicos de rinitis, incremento en la salivación, miosis, lagrimeo, diaforesis, disnea e hiperperistaltismo intestinal que puede ocasionar calambres abdominales. La diarrea temprana y otros síntomas colinérgicos se pueden evitar o disminuir con la administración de atropina.La diarrea tardía (se presenta generalmente más de 24 horas después de la administración) puede poner en riesgo la vida, ya que se puede prolongar ocasionando deshidratación, desequilibrio hidroelectrolítico o sepsis. La diarrea tardía debe ser tratada rápidamente con loperamida. Los pacientes con diarrea se deben monitorear cuidadosamente, se les debe administrar reemplazo de fluidos y electrólitos si están deshidratados, así como antibiótico si desarrollan íleo, fiebre o neutropenia severa, después del primer tratamiento. La quimioterapia semanal subsecuente se debe retrasar en los pacientes, hasta que regresen a su función intestinal normal previa al tratamiento y que ésta se mantenga por al menos 24 horas sin necesidad de medicamentos antidiarreicos.Náusea y vómito: El irinotecan es emetogénico. Las náuseas y el vómito pueden ser severos y usualmente ocurren durante o poco después de la infusión del irinotecan. Se recomienda que los pacientes reciban premedicación con agentes antieméticos.Los agentes antieméticos se administrarán en el día del tratamiento, empezando por lo menos 30 minutos antes de la administración del irinotecan.Además, los médicos deben considerar prescribir a sus pacientes un régimen antiemético para uso subsiguiente según se requiera.Pacientes con vómito asociado a diarrea tardía deben ser hospitalizados tan pronto sea posible para tratamiento. Deben evitar el uso de fármacos con propiedades laxantes debido a la posible exacerbación de la diarrea.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: El irinotecan es teratogénico en ratas y conejos.El irinotecan puede causar daño fetal cuando se administra a una mujer embarazada.No se han realizado estudios adecuados y bien controlados con irinotecan en mujeres embarazadas. Si el fármaco se usa durante el embarazo, o si la paciente se embaraza mientras está recibiendo este fármaco, debe ser advertida del peligro potencial para el feto. Las mujeres con probabilidades de embarazo deben ser advertidas de evitar embarazarse mientras reciben tratamiento con irinotecan.Lactancia: En ratas se detectó radiactividad en la leche dentro de 5 minutos de la administración intravenosa de irinotecan marcado radiactivamente y se concentró hasta 65 veces a las 4 horas después de la administración, en comparación con las concentraciones plasmáticas.Como muchos fármacos se excretan en la leche humana y dado el potencial de reacciones adversas severas en los lactantes, se recomienda interrumpir la lactancia cuando se recibe terapia con irinotecan.
Reacciones secundarias y adversas: Las hospitalizaciones debidas a efectos adversos severos, relacionados o no con la administración de irinotecan se presentaron en al menos 60% de los pacientes que recibieron el fármaco y el 8% de ellos interrumpieron el tratamiento debido a la severidad de las mismas. Se han recolectado y analizado exhaustivamente los datos de los eventos adversos obtenidos en el programa de estudios clínicos de cáncer colorrectal metastásico recurrente o progresivo después de terapia con 5-FU (segunda línea). Se espera que los eventos adversos para otras indicaciones sean similares a los eventos adversos de la terapia de segunda línea para el cáncer colorrectal.Los efectos adversos grado 1 a 4 (Clasificación del National Cancer Institute [NCI]) relacionados con el fármaco, reportados en más del 10% de los pacientes en los estudios clínicos fueron diarrea tardía, náusea, vómito, diarrea temprana, anorexia, estomatitis, astenia, calambres/dolor abdominal, pérdida de peso, deshidratación, alopecia y eventos tromboembólicos.A continuación se enlistan los eventos adversos severos de grados 3 o 4 (del NCI), reportados en estudios clínicos utilizando los esquemas de dosificación semanal y de una vez cada 3 semanas. Observados en más del 10% de los pacientes: diarrea tardía, náusea, leucopenia, neutropenia, alopecia. En 1 al 10%: Infección, vómito, diarrea temprana, constipación, anorexia, mucositis, anemia, trombocitopenia, astenia, fiebre, dolor, deshidratación, bilirrubinemia, creatinina elevada, hipovolemia y disnea.En menos del 1%: Sepsis, obstrucción intestinal, íleo, candidiasis gastrointestinal, escalofríos, astenia, adinamia, pérdida de peso, aumento de fosfatasa alcalina, aumento de TGP, hipopotasemia, hipomagnesemia, erupción cutánea, parestesias, calambres, marcha anormal, confusión, cefalea, hipotensión, trastornos cardiovasculares, infección del tracto urinario, mastalgias.
Interacciones medicamentosas y de otro género: Agentes bloqueadores neuromusculares: Debido a que irinotecan tiene actividad anticolinesterasa, los medicamentos con actividad anticolinesterasa pueden prolongar los efectos de bloqueo neuromuscular de suxametonio.Agentes antineoplásicos: Se espera que los efectos adversos del irinotecan, como mielosupresión y diarrea, sean exacerbados por otros agentes antineoplásicos con un perfil similar de efectos adversos.Dexametasona: Se ha reportado linfocitopenia en los pacientes tratados con irinotecan, y es posible que la administración de dexametasona como profilaxis antiemética pueda tener una mayor probabilidad de linfocitopenia. Sin embargo, no se han observado infecciones oportunistas, y no se ha atribuido ninguna complicación específicamente a la linfocitopenia. Se ha observado hiperglucemia en los pacientes con historia de diabetes mellitus o evidencia de intolerancia a la glucosa antes de la administración del irinotecan. Es probable que en algunos pacientes la dexametasona, administrada como profilaxis antiemética, contribuya a la hiperglucemia.Laxantes: Se espera que el uso de laxantes durante la terapia empeore la incidencia o severidad de la diarrea.Diuréticos: El irinotecan puede inducir deshidratación secundaria a vómito y/o diarrea. El médico puede suspender los diuréticos durante el tratamiento con irinotecan y durante los periodos de vómito y diarrea activos.Anticonvulsivantes: La administración conjunta de medicamentos anticonvulsivantes (carbamazepina, fenobarbital y fenitoína) conducen a una exposición disminuida del metabolito activo SN-38. Debe tomarse en consideración para el inicio o sustitución de anticonvulsivantes no enzimáticos por lo menos una semana antes del inicio de la terapia con irinotecan en pacientes que requieren tratamiento anticonvulsivante.Ketoconazol: La depuración de irinotecan es ampliamente reducida en pacientes que reciben conjuntamente ketoconazol conduciendo a una exposición incrementada a SN-38. Ketoconazol debe ser discontinuado por lo menos 1 semana antes del inicio de la terapia con irinotecan y no debe ser administrado durante la terapia con irinotecan.
Alteraciones en los resultados de pruebas de laboratorio: No existen interacciones conocidas entre las pruebas de laboratorio y el irinotecan; sin embargo, se recomienda la monitorización cuidadosa de la cuenta de leucocitos, hemoglobina y cuenta plaquetaria antes de cada dosis de ETONIRI®.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Toxicología: La toxicidad aguda de irinotecan en animales es mostrada más adelante. La toxicidad subaguda de irinotecan mostró afectos tisulares con una rápida proliferación celular (médula ósea, epitelio intestinal, timo, bazo, nódulos linfáticos y testículos). Mutagénesis/carcinogénesis: No se han llevado a cabo estudios a largo plazo con irinotecan. Sin embargo, se han administrado dosis intravenosas a las ratas de 2 mg/kg o 25 mg/kg de irinotecan una vez por semana por 13 semanas (en estudios separados la dosis de 25 mg/kg produjo una Cmáx. de irinotecan y una ABC 7 veces y 1.3 veces los valores respectivos en pacientes a los que se les administró 125 mg/m²) y se recuperaron después de 91 semanas. Ni el irinotecan ni el SN-38 fueron mutagénicos en los ensayos in vitro. Sin embargo, en estudios in vivo en células de hámster chinos, el irinotecan produjo un incremento significativo en la incidencia de aberraciones cromosomales dependiente de la concentración. Adicionalmente, en un ensayo in vivo con ratones, una sola dosis intraperitoneal de irinotecan sobre el intervalo de la dosis de 2.5 a 200 mg/kg causó un incremento significativo dependiente de la dosis en la formación de eritrocitos policromáticos y una disminución en la proporción de reticulocitos/eritrocitos en las células de la médula ósea. Reproducción: No se han observado efectos adversos significativos en la fertilidad y la reproductividad general después de la administración intravenosa de irinotecan en dosis de hasta 6.0 mg/kg/día en ratas. Sin embargo, la atrofia de los órganos reproductores masculinos se ha observado después de la administración de dosis múltiples diarias de irinotecan, tanto en roedores como en perros.La radiactividad relacionada con el irinotecan marcado con C14 cruza la placenta de las ratas después de la administración intravenosa de 10 mg/kg, que corresponden a valores de 125 mg/m² administrados en pacientes, y en conejos a 6 mg/kg/día (aproximadamente la mitad de la dosis humana recomendada semanalmente). Los efectos teratogénicos incluyen una variedad de efectos externos, viscerales y esqueléticos. El irinotecan administrado en ratas madres para el periodo seguido a la organogénesis a través de la lactancia a dosis de 6 mg/kg/día causó disminución en la capacidad de aprendizaje y disminuyó los pesos corporales de las hembras en el nacimiento.
Dosis y vía de administración: ETONIRI® se debe administrar exclusivamente en infusión intravenosa durante 30 a 90 minutos, debe diluirse previo a la infusión en una solución de dextrosa al 5% para inyección, o en cloruro de sodio a 0.9% para inyección, hasta obtener una concentración final de 0.12 a 2.8 mg/ml. Se deben observar medidas de seguridad en el manejo y preparación de las soluciones que contienen agentes antineoplásicos como es el caso en el manejo de ETONIRI®, recomendándose el uso de guantes. En caso de que la solución entre en contacto con la piel o mucosas, se debe lavar de inmediato con abundante agua y jabón.Una vez hecha la mezcla, la solución es física y químicamente estable por 24 horas a temperatura ambiente.Las soluciones diluidas en dextrosa al 5% para inyección, almacenadas en refrigeración y protegidas de la luz, son física y químicamente estables durante 48 horas. No se recomienda la refrigeración de las mezclas preparadas con cloruro de sodio al 0.9% para inyección, debido a una baja y esporádica incidencia de partículas visibles.Debido a la posible contaminación durante la dilución, es aconsejable usar la mezcla dentro de 24 horas en refrigeración o dentro de 6 horas si se mantiene a temperatura ambiente.Se debe evitar congelar los viales o las mezclas de irinotecan, en vista de que puede causar la precipitación del principio activo. No se deben adicionar otros fármacos en la solución para infusión. Esquema de dosificación como agente único: Esquema de dosificación semanal: La dosis inicial recomendada de ETONIRI® solo es de 125 mg/m². Se puede considerar una dosis menor (por ejemplo, 100 mg/m² para los pacientes con cualquiera de las siguientes condiciones: edad de 65 años o más, radioterapia extensa previa, estado de condición física de 2 (ECOG), niveles aumentados de bilirrubina o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, que comprenden el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas.Esquema de dosificación una vez cada 2 semanas: La dosis inicial usual recomendada de ETONIRI® es de 250 mg/m² cada 2 semanas mediante infusión intravenosa. Se puede considerar una dosis inicial menor (por ejemplo, 200 mg/m²) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2 (ECOG), niveles aumentados de bilirrubina o cáncer gástrico. Esquema de dosificación una vez cada 3 semanas: La dosis inicial usual recomendada de ETONIRI® para el esquema de dosificación una vez cada 3 semanas, es de 350 mg/m². Se puede considerar una dosis inicial menor (por ejemplo, 300 mg/m²) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2 (ECOG), niveles aumentados de bilirrubina o cáncer gástrico. Esquema de dosificación en combinación: ETONIRI® en combinación con 5-fluorouracilo (5-FU) y leucovorina: Se recomienda el uso de ETONIRI® en combinación con 5-FU y leucovorina en los pacientes con cáncer colorrectal metastásico.La dosis inicial recomendada es de 125 mg/m² de ETONIRI®, 500 mg/m² de 5-FU y 20 mg/m² de leucovorina. Se pueden considerar dosis iniciales menores de ETONIRI® (por ejemplo, 100 mg/m²) y 5-FU (por ejemplo, 400 mg/m²) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad y mayores, radioterapia extensa previa, estado de condición física de 2 (ECOG), niveles aumentados de bilirrubina o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, que comprenden el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas. ETONIRI® en combinación con cisplatino: ETONIRI® se ha estudiado en combinación con cisplatino para el cáncer pulmonar de células no pequeñas y de células pequeñas, cáncer cervical, cáncer gástrico y cáncer esofágico. Este régimen se puede usar en el tratamiento de otros cánceres indicados, excepto para el cáncer colorrectal (véase Indicaciones terapéuticas).La dosis inicial recomendada es de 65 mg/m² de ETONIRI® y 30 mg/m² de cisplatino. Se puede considerar una dosis inicial menor de ETONIRI® (por ejemplo, 50 mg/m²) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2 (ECOG), niveles aumentados de bilirrubina o cáncer gástrico.El tratamiento se debe administrar en ciclos repetidos de 6 semanas, comprendiendo el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas.Duración del tratamiento: Para los regímenes de ETONIRI® solo y en combinación, se puede continuar indefinidamente el tratamiento con ciclos adicionales de ETONIRI® en los pacientes que logran una respuesta tumoral o en los pacientes cuyo cáncer permanece estable.Los pacientes deben ser vigilados cuidadosamente para detectar toxicidad y se debe suspender la terapia si ocurre toxicidad evidente que no responda a la modificación de la dosis y al cuidado rutinario de soporte. Poblaciones especiales: Pacientes con insuficiencia hepática: En pacientes con disfunción hepática se deberá ajustar la dosis de acuerdo a las concentraciones de bilirrubinas y de las enzimas hepáticas TGP y TGO. Pacientes con insuficiencia renal: Estudios en esta población no han sido realizados (véase Propiedades farmacocinéticas; Farmacocinética en poblaciones especiales). Por lo tanto, deben tomarse precauciones en pacientes con función renal deteriorada. Irinotecan no es recomendado para el uso en pacientes en diálisis.Recomendaciones para la modificación de la dosis: Ver tabla siguiente (7) se describen las modificaciones recomendadas de las dosis durante un ciclo de terapia y al inicio de cada ciclo subsiguiente de terapia con irinotecan solo. Estas recomendaciones se basan en las toxicidades comúnmente observadas con la administración de irinotecan. Para las modificaciones al inicio de un ciclo subsiguiente de terapia, la dosis de irinotecan se debe disminuir con relación a la dosis inicial del ciclo previo. En la tabla 8 se describen las modificaciones recomendadas de las dosis de irinotecan y 5-FU durante un ciclo de terapia y al inicio de cada ciclo subsiguiente de terapia con irinotecan, 5-FU y leucovorina. En la tabla 9 se describen las modificaciones recomendadas de las dosis de irinotecan y cisplatino para el inicio de cada ciclo de terapia, y en la tabla 10 se describen las recomendaciones para las modificaciones de las dosis durante un ciclo de terapia.Todas las modificaciones de las dosis se deben basar en la peor toxicidad precedente.No se debe iniciar un nuevo ciclo de terapia hasta que la toxicidad haya desaparecido a un grado 2 del NCI o menor.El tratamiento se puede retrasar 1 a 2 semanas para permitir que el paciente se recupere de la toxicidad relacionada con el tratamiento. Si el paciente no se ha recuperado, se debe considerar interrumpir el irinotecan.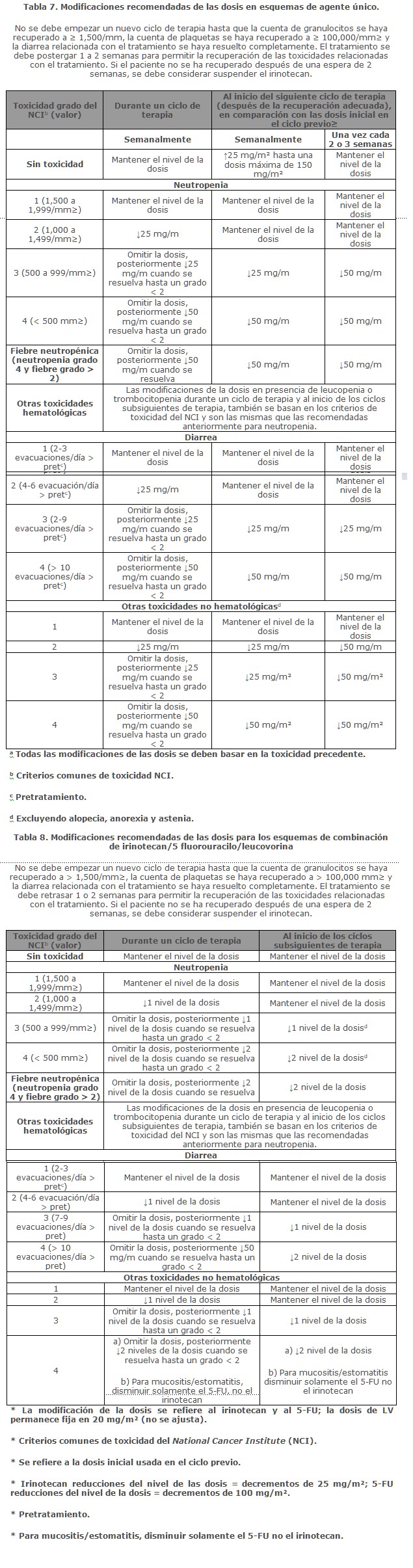
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Se han administrado dosis únicas de hasta 750 mg/m² de irinotecan a pacientes con diversos cánceres. Los eventos adversos en estos pacientes fueron similares a los reportados con las dosis y regímenes recomendados. Las reacciones adversas más significativas fueron neutropenia y diarrea severa. Se debe instituir el cuidado máximo de soporte para prevenir la deshidratación debida a diarrea y tratar cualquier complicación infecciosa. No hay antídoto conocido para la sobredosificación con irinotecan.
Presentaciones: ETONIRI® caja con un frasco ámpula con 100 mg/5 ml.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente entre 15 y 30°C. Se recomienda que el vial se mantenga en su empaque de cartón hasta el momento de su uso.No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado. Si no se administra todo el producto deséchese el sobrante. Inspeccionar el contenido del vial para detectar partículas y repetir la inspección al tener el producto en la jeringa. Las soluciones diluidas en dextrosa al 5% se deben conservar en refrigeración entre 2°C a 8°C durante 48 horas. Las soluciones diluidas en cloruro de sodio al 0.9% se deben conservar a temperatura ambiente hasta 24 horas.
Leyendas de protección: Vía de administración: Exclusivamente por vía intravenosa y previamente diluido en solución de dextrosa al 5% o de cloruro de sodio al 0.9%. Dosis: La que el médico señale. Su venta requiere receta médica. No se deje al alcance de los niños. Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología y con experiencia en quimioterapia antineoplásica. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. Consérvese a temperatura ambiente a no más de 30°C, no se congele. Las soluciones diluidas en dextrosa al 5% se deben conservar en refrigeración entre 2 a 8°C durante 48 horas. Las soluciones diluidas en cloruro de sodio al 0.9% se deben conservar a temperatura ambiente hasta 24 horas. Léase instructivo anexo. No se use en el embarazo y la lactancia. Medicamento de alto riesgo.
Nombre y domicilio del laboratorio: Hecho en México por: Dinafarma, S.A. de C.V. Circuito Nemesio Diez Riega No. 10, Col. Parque Industrial el Cerrillo II, C.P. 52000, Lerma, Estado de México, México. Distribuido por: LABORATORIOS SANFER, S.A. de C.V. Hormona No. 2-A,San Andrés Atoto, C.P. 53500 Naucalpan de Juárez, Estado de México, México.
Número de registro del medicamento: 390M2008, SSA IV