ETURION
SANFER
Denominación genérica: Atorvastatina Cálcica.
Forma farmacéutica y formulación: Tabletas.
Cada tableta contiene: Atorvastatina cálcica* equivalente a 10 mg, 20 mg, 40 mg y 80 mg de atorvastatina Excipiente, c.b.p. 1 tableta. * Trihidratada.
Indicaciones terapéuticas: Atorvastatina está indicada como auxiliar a la dieta para el tratamiento de pacientes con triglicéridos, apolipoproteína B, coIesterol-LDL (C-LDL) y colesterol total elevados y para incrementar el colesterol HDL (C-HDL) en pacientes con hipercolesterolemia primaria (hipercolesterolemia familiar y no familiar heterocigótica), hiperlipidemia combinada (mixta) (Fredrickson tipos IIa y IIb), aumento de niveles de triglicéridos séricos (Fredrickson tipo IV), y para pacientes con disbetalipoproteinemia (Fredrickson tipo III) que no responden adecuadamente a la dieta. Atorvastatina también está indicada para la reducción de colesterol total y colesterol-LDL en pacientes con hipercolesterolemia familiar homocigota. Prevención de complicaciones cardiovasculares (CV): En pacientes sin enfermedad cardiovascular clínicamente evidente, y con o sin dislipidemia, pero con múltiples factores de riesgo para enfermedad cardiaca coronaria tales como tabaquismo, hipertensión, diabetes, C-HDL bajo, o un historial familiar de enfermedad cardiaca coronaria en etapa temprana, atorvastatina está indicada para: Reducir el riesgo de enfermedad cardiaca coronaria fatal e infarto del miocardio no fatal. Reducir el riesgo de evento vascular cerebral. Reducir el riesgo de procedimientos de revascularización y angina de pecho. En pacientes con enfermedad coronaria evidente, atorvastatina está indicada para: Reducir el riesgo de infarto del miocardio no fatal. Reducir el riesgo de evento vascular cerebral fatal y no fatal. Reducir el riesgo de procedimientos de revascularización. Reducir el riesgo de hospitalización por insuficiencia cardiaca. Reducir el riesgo de angina. Pacientes pediátricos (10-17 años de edad): Atorvastatina está indicada como auxiliar de la dieta para reducir los niveles de apo B, C-LDL, y C total en niños y niñas luego de la menarca, de 10 a 17 años de edad, con hipercolesterolemia familiar heterocigótica si después de un adecuado manejo con terapia dietética se presentan los siguientes hallazgos: a. C-LDL permanece ≥ 190 mg/dl o b. C-LDL permanece ≥ 160 mg/dl y: Existen un historial familiar positivo de enfermedad cardiovascular prematura o Dos o más factores de riesgo CV están presentes en el paciente pediátrico.
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Absorción: La atorvastatina es rápidamente absorbida después de su administración oral; las concentraciones plasmáticas máximas ocurren dentro de una a dos horas. El grado de absorción y las concentraciones plasmáticas de atorvastatina aumentan en proporción a la dosis de atorvastatina. Las tabletas de atorvastatina son de 95% a 99% biodisponibles en comparación con las soluciones. La biodisponibilidad absoluta de atorvastatina es aproximadamente 14% y la disponibilidad sistémica de la actividad inhibitoria de HMG-CoA reductasa es aproximadamente 30%. La disponibilidad sistémica baja se atribuye a la depuración presistémica en la mucosa gastrointestinal y/o al metabolismo hepático en su primer paso. A pesar de que los alimentos disminuyen la tasa y el grado de absorción del fármaco en aproximadamente 25% y 9%, respectivamente, según se evalúa por Cmáx. y Área Bajo la Curva (ABC), la reducción de C-LDL es similar, ya sea que atorvastatina se administre con o sin alimentos. Las concentraciones plasmáticas de atorvastatina son menores (aproximadamente 30% para Cmáx. y ABC) tras administración farmacológica por la noche en comparación con la mañana. Sin embargo, la reducción de C-LCL es la misma sin importar la hora del día de la administración del fármaco (véase Dosis y vía de administración). Distribución: El volumen medio de distribución de atorvastatina es aproximadamente 381 litros. Atorvastatina tiene un enlace ≥ 98% con las proteínas plasmáticas. Una proporción de eritrocitos/plasma de aproximadamente 0.25 indica una penetración deficiente del fármaco en los eritrocitos. Metabolismo: Atorvastatina es extensamente metabolizada a derivados orto y parahidroxilados y a varios productos de beta-oxidación. La inhibición in vitro de HMG-CoA reductasa por medio de metabolitos orto y parahidroxilados es equivalente a la de atorvastatina. Aproximadamente 70% de la actividad inhibitoria circulante para HMG-CoA reductasa se atribuye a los metabolitos activos. Los estudios in vitro sugieren la importancia del metabolismo de atorvastatina mediante citocromo P-450 3A4 hepático, consistente con el aumento de las concentraciones plasmáticas de atorvastatina en humanos tras coadministración con eritromicina, un inhibidor conocido de esta isoenzima. Los estudios in vitro también indican que atorvastatina es un inhibidor débil del citocromo P-450 3A4. La coadministración de atorvastatina no produjo un efecto clínicamente significativo en las concentraciones plasmáticas de terfenadina, un compuesto predominantemente metabolizado por citocromo P-450 3A4; por lo tanto, es improbable que atorvastatina altere significativamente la farmacocinética de otros sustratos del citocromo P-450 3A4 (véase Interacciones medicamentosas y de otro género). En animales, el metabolito orto-hidroxi sufre glucuronidación adicional. Excreción: Atorvastatina y sus metabolitos son eliminados principalmente en la bilis tras metabolismo hepático y/o extrahepático; sin embargo, no parece que el fármaco sufra recirculación enterohepática. La vida media de eliminación plasmática promedio de atorvastatina en humanos es aproximadamente 14 horas, pero la vida media de la actividad inhibitoria para HMG-CoA reductasa es de 20 a 30 horas debido a la contribución de los metabolitos activos. Menos del 2% de una dosis de atorvastatina se recupera en la orina tras la administración oral. Poblaciones especiales: Edad avanzada: Las concentraciones plasmáticas de atorvastatina son mayores (aproximadamente 40% para Cmáx. y 30% para ABC) en sujetos saludables de edad avanzada (edad ≥ 65 años) que en adultos jóvenes. El estudio ACCESS específicamente evaluó a pacientes de edad avanzada en relación al alcance de sus metas del tratamiento NCEP. El estudio incluyó a 1,087 pacientes menores de 65 años de edad, 815 pacientes mayores de 65 años de edad, y 185 pacientes mayores de 75 años de edad. No se observaron diferencias en seguridad, eficacia o logro de la meta del tratamiento de lípidos entre pacientes de edad avanzada y la población general. Niños: No se han realizado estudios de farmacocinética en la población pediátrica. Género: Las concentraciones plasmáticas de atorvastatina son diferentes en mujeres (aproximadamente 20% más elevadas para Cmáx. y 10% más bajas para ABC) que en los hombres. Sin embargo, no hubo diferencias clínicamente significativas en efectos de lípidos entre hombres y mujeres. Insuficiencia renal: La enfermedad renal no tiene influencia sobre las concentraciones plasmáticas o sobre los efectos de lípidos de atorvastatina. Por lo tanto, no es necesario el ajuste de dosis en pacientes con disfunción renal (véase Dosis y vía de administración). Hemodiálisis: Debido a que no se han realizado estudios en pacientes con enfermedad renal en etapa terminal, no se espera que la hemodiálisis mejore significativamente la depuración de atorvastatina debido al enlace extenso del fármaco a las proteínas plasmáticas. Insuficiencia hepática: Las concentraciones plasmáticas de atorvastatina están notablemente aumentadas (aproximadamente 16 veces en Cmáx. y 11 veces en ABC) en pacientes con enfermedad hepática alcohólica crónica (Child-Pugh B) (véase Contraindicaciones). Interacciones medicamentosas: A continuación se resumen el efecto de los medicamentos administrados conjuntamente sobre la farmacocinética de la atorvastatina, así como el efecto de la atorvastatina sobre la farmacocinética de los medicamentos administrados conjuntamente (véase Precauciones generales e Interacciones medicamentosas y de otro género). Efecto de los medicamentos administrados conjuntamente sobre la farmacocinética de la atorvastatina: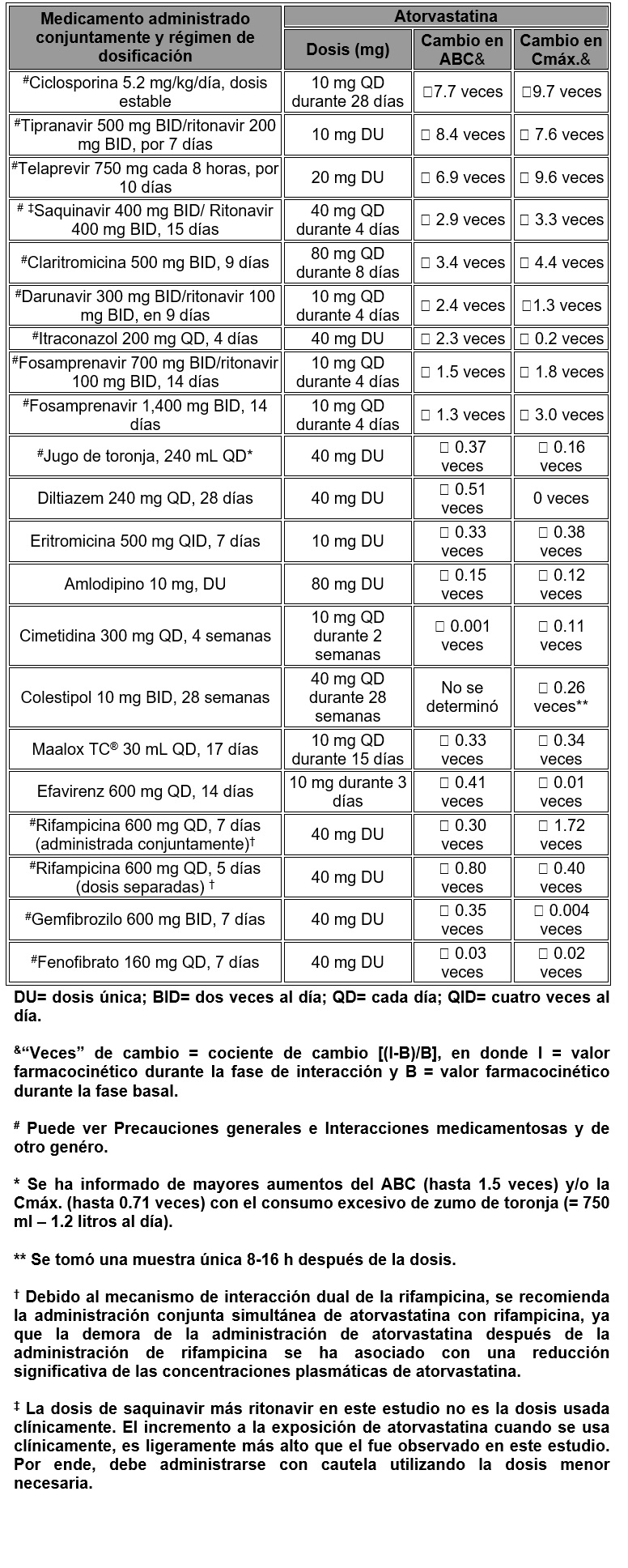
Efecto de la atorvastatina sobre la farmacocinética de los medicamentos administrados conjuntamente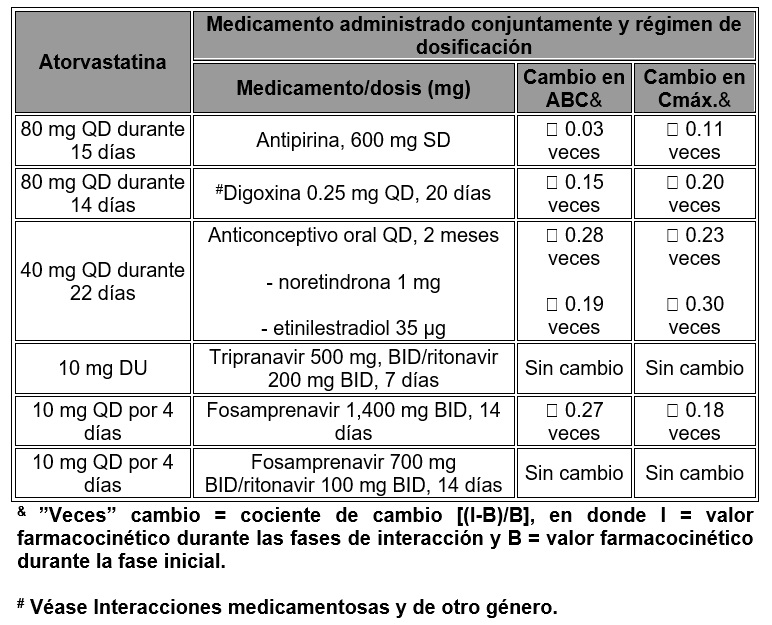
Propiedades farmacodinámicas: Atorvastatina calcica es un agente sintético para la disminución de los lípidos, inhibidor de la 3-hidroxi-3-metilglutaril-coenzima A (HMG-CoA) reductasa. Esta enzima cataliza la conversión de HMG-CoA a mevalonato, un paso temprano y limitante de la velocidad en la biosíntesis del colesterol. La fórmula empírica de atorvastatina cálcica es (C33H34FN2O5)2 Ca 3H20 con peso molecular de 1209.42 y cuya fórmula estructural es: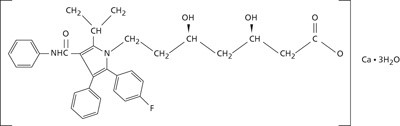
La atorvastatina cálcica es un polvo cristalino blanco a grisáceo particularmente insoluble en soluciones acuosas de pH 4 e inferior, muy poco soluble en agua destilada, amortiguador de fosfato de pH 7.4 y acetonitrilo, ligeramente soluble en etanol y por completo soluble en metanol. Mecanismo de acción: La atorvastatina es un inhibidor selectivo competitivo de la reductasa de HMG-CoA, enzima limitante de la velocidad de la reacción que convierte en la 3-hdroxi-3-metilglutaril-co enzima A en mevalonato, un precursor de los esteroides, incluido el colesterol. En pacientes con hipercolesterolemia familiar (FH) homocigota y heterocigota, formas no familiares de hipercolesterolemia y dislipidemia mixta, atorvastatina disminuye el C-total (colesterol total), C-LDL (colesterol de lipoproteínas de baja densidad) y apo B (apolipoproteína B). Atorvastatina también disminuye C-VLDL (colesterol de lipoproteínas de muy baja densidad) y TG (triglicéridos) y produce aumentos variables de C-HDL (colesterol de lipoproteínas de alta densidad). La atorvastatina disminuye las concentraciones de colesterol plasmático y lipoproteínas por inhibición de la reductasa de HMG-CoA reductasa y la síntesis de colesterol en el hígado, y aumenta el número de receptores hepáticos de LDL en la superficie celular para una captación y un catabolismo mayor LDL. La torvastatina disminuye la producción y el número de partículas de LDL. Causa un incremento intenso y sostenido de la actividad del receptor de LDL acoplado con un cambio benéfico en la calidad de las partículas circulantes de LDL. La atorvastatina es eficaz para disminuir LDL en pacientes con hipercolesterolemia familiar homocigota, una población que no ha respondido normalmente a los medicamentos de disminución de lípidos. La atorvastatina y algunos de sus metabolitos tienen actividad farmacológica en seres humanos. El principal sitio de acción de la atorvastatina es el hígado, que es sitio de síntesis de colesterol y depuración de LDL más importante. La disminución de C-LDL se correlaciona mejor con la dosis de fármaco que con su concentración sistémica. La individualización de la dosis de uso del fármaco debería basarse en la respuesta terapéutica (véase Dosis y vía de administración). En un estudio de dosis-respuesta, la atorvastatina (10-80 mg) disminuyó el C-total (30%-46%), el C-LDL (41%-61%), la apo B (34%-50%) y los TG (14%-33%), resultados consistentes en pacientes con hipercolesterolemia familiar heterocigota, formas no familiares de hipercolesterolemia, e hiperlipidemia mixta, incluidos aquellos con diabetes mellitus no insulinodependiente. En pacientes con hipertrigliceridemia aislada, la atorvastatina disminuye C-total, C-LDL, C-VLDL, apo B, TG y colesterol no-HDL, y aumenta C-HDL. En pacientes con disbetalipoproteinemia, la atorvastatina disminuye C-IDL (colesterol de lipoproteínas de densidad intermedia). En pacientes con hiperlipoproteinemia de Fredrickson tipos IIa y IIb conjuntados de 24 estudios controlados, el incremento porcentual promedio respecto de la basal en C-HDL para atorvastatina (10-80 mg) fue de 5.1 a 8.7% en una forma no relacionada con la dosis. Adicionalmente, el análisis de esos datos acumulados demostró un decremento en las razones C-total/C-HDL C-LDL/C-HDL significativamente relacionado con la dosis, con variación de -29 a -44% y -37 a -55%, respectivamente. Los efectos de atorvastatina sobre eventos isquémicos y mortalidad total se estudiaron en el estudio Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL), estudio multicéntrico aleatorio, doble ciego, con placebo, controlado, de vigilancia de 3,086 pacientes con síndromes coronarios agudos; angina inestable o infarto miocárdico sin onda Q. Los pacientes se trataron con las medidas estándar, incluida la dieta, y atorvastatina a dosis de 80 mg diarios o placebo durante un promedio de 16 semanas. Las cifras finales de C-LDL, C-total, C-HDL y TG fueron 72, 147, 48, 139 mg/dl, en ese orden, en el grupo con atorvastatina, y de 135, 217, 46 y 187 mg/dl, respectivamente, en el grupo con placebo. Atorvastatina disminuyó significativamente el riesgo de eventos isquémicos y muerte en un 16%. El riesgo de rehospitalización por angina de pecho, con datos demostrados de isquemia miocárdica, disminuyó significativamente en 26%. Atorvastatina aminoró el riesgo de eventos isquémicos y muerte en un grado similar dentro de la variación del C-LDL basal. Además, atorvastatina disminuyó el riesgo de eventos isquémicos y muerte en grados similares en pacientes con infarto del miocardio (IM) sin onda Q y angina inestable, así como en hombres y mujeres y en pacientes ≥ 65 y > 65 años de edad. Prevención de las complicaciones cardiovasculares: En el estudio Anglo-Scandinavian Cardiac Outcomes Lipid Lowering Arm (ASCOT-LLA) se valoró el efecto de atorvastatina sobre la enfermedad coronaria fatal y no fatal en 10,305 pacientes hipertensos de 40 a 80 años de edad (promedio de 63 años) sin infarto del miocardio previo y con cifras de colesterol total (TC) < 6.5 mmol/l (251 mg/dl). Adicionalmente, todos los pacientes tenían al menos tres de los siguientes factores de riesgo cardiovascular: género masculino, edad > 55 años, tabaquismo, diabetes, antecedente de enfermedad coronaría en un pariente de primer grado, CT: C-HDL > 6 mmol/L, enfermedad vascular periférica, hipertrofia ventricular izquierda, evento vascular cerebral previo, una anomalía específica en el EKG, proteinuria/albuminuria. En este estudio doble ciego controlado con placebo se trató a los pacientes con antihipertensivos (metas de presión arterial < 140/90 mm Hg para los no diabéticos y < 130/80 mm Hg para los diabéticos) y atorvastatina 10 mg diarios (n = 5,168) o placebo (n = 5,137). Como el efecto del tratamiento con atorvastatina en comparación con placebo rebasó el umbral de significancia durante un análisis interino del ASCOT-LLA, el estudio concluyó en 3.3 años y no en los cinco años planeados inicialmente. Adicionalmente, la presión arterial fue bien controlada y similar en pacientes asignados para recibir atorvastatina y placebo. Esos cambios persistieron durante el periodo de tratamiento. La atorvastatina disminuyó significativamente las tasas de los siguientes eventos: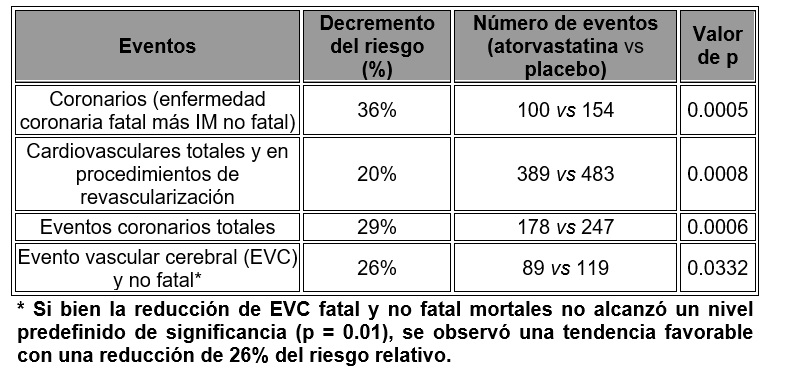
La mortalidad total y la mortalidad cardiovascular no han sido reducidas significativamente, aunque se observó una tendencia favorable. En el estudio Collaborative Atorvastatin Diabetes (CARDS) se valoró el efecto de atorvastatina sobre la enfermedad cardiovascular fatal y no fatal, en 2,838 pacientes con diabetes de tipo II de 40 a 75 años de edad sin antecedente de enfermedad cardiovascular y con LDL ≥ 4.14 mmol/L (160 mg/dl) y TG ≤ 6.78 mmol/L (600 mg/dl). Adicional mente todos los pacientes tenían al menos uno de los siguientes factores de riesgo: hipertensión, tabaquismo actual, retinopatía, microalbuminuria o macroalbuminuria. En este estudio aleatorio, doble ciego, multicéntrico, placebo controlado, se trató a los pacientes con atorvastatina 10 mg diarios (n = 14,28) o placebo (n = l,410) durante un promedio de 3.9 años de vigilancia de la evolución. Como el efecto del tratamiento con atorvastatina sobre el criterio de valoración primario alcanzó las reglas predefinidas y detener el estudio en cuanto a eficacia, el CARDS concluyó dos años antes de lo previsto. El efecto de disminución del riesgo absoluto y relativo por atorvastatina es el siguiente: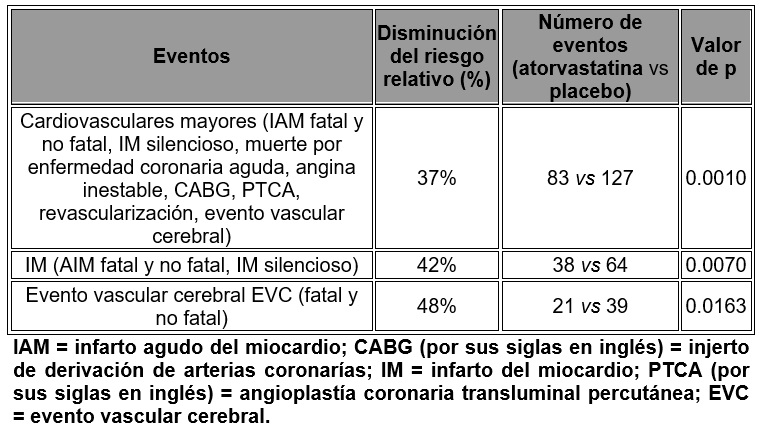
No hubo evidencia de una diferencia en el efecto del tratamiento por género, edad o cifra basal de C-LDL del paciente. Se observó una disminución del riesgo relativo de muerte de 27% (82 muertes en el grupo con placebo en comparación con 61 en el de tratamiento), con significancia estadística limítrofe (p = 0.0592). La incidencia global de eventos adversos o eventos adversos serios fue similar entre los grupos de tratamiento. Aterosclerosis: En el estudio Reversing Atherosclerosis with Aggressvive Lipid-Lowering (REVERSAL), se determinó el efecto de atorvastatina 80 mg y pravastatina 40 mg sobre la aterosclerosis coronaria por ultrasonografía intravascular (IVUS, por sus siglas en inglés), durante la angiografía, en pacientes con enfermedad coronaria. En ese estudio clínico, doble ciego, aleatorio, multicéntrico, controlado, se hizo IVUS basal y a los 18 meses en 502 pacientes. En el grupo de atorvastatina (n = 253), el cambio porcentual promedio respecto de la basal en el volumen total del ateroma (principal criterio de estudio) fue de -0.4% (p = 0.98) en el grupo de atorvastatina y +2.7% (p = 0.001) en el de pravastatina (p = 249). Al compararlos con pravastatina, los efectos de atorvastatina fueron estadísticamente significativos (p = 0.02). En el grupo de atorvastatina, el C-LDL disminuyó hasta un promedio de 2.04 mmol/L ± 0.8 (78.9 mg/dl ± 30) respecto a la basal de 3.89 mmol/L ± 0.7 (150 mg/dl ± 28), y en el grupo de pravastatina, el C-LDL disminuyó un promedio hasta una media de 2.85 mmol/L ± 0.7 (110 mg/dl ± 26) respecto a la basal de 3.89 mmol/L ± 0.7 (150 mg/dl ± 26) (p < 0.0001). Atorvastatina también disminuyó significativamente el colesterol total promedio en 34.1% (pravastatina: -18.4%, p < 0.001), la cifras promedio de triglicéridos por 20% (pravastatina: -6.8%, p < 0.0009) y la apolipoproteína B promedio por 39.1% (pravastatina: -22.0%, p < 0.0001). Atorvastatina aumentó el C-HDL promedio de 2.9% (pravastatina: + 5.6% p = NS). Hubo una disminución promedio de 36.4% en la proteína C reactiva (PCR) en el grupo con atorvastatina, en comparación con 5.2% en el de pravastatina (p < 0.0001). Los perfiles de seguridad y tolerabilidad de los dos grupos de tratamiento fueron comparables. Evento vascular cerebral (EVC) recurrente: En el estudio Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL), se valoró el efecto de la dosis de atorvastatina de 80 mg diarios o placebo sobre los eventos vasculares cerebrales en 4,731 pacientes con un evento vascular cerebral o un ataque isquémico transitorio (ATI) en los seis meses precedentes y sin antecedentes de enfermedad coronaria. Los pacientes fueron 60% hombres, de 21 a 92 años de edad (media de 63) y tenían una cifra de LDL basal promedio de 133 mg/dl (3.4 mmol/L). La media de C-LDL fue de 73 mg/dl (1.9 mmol/L) durante, el tratamiento con atorvastatina y 129 mg/dl (3.3 mmol/L) durante el uso de placebo. El tiempo promedio de vigilancia de la evolución fue de 4.9 años. Atorvastatina a dosis de 80 mg disminuyó el riesgo del punto final primario, de evento vascular cerebral fatal o no fatal en 15% (HR 0.85; IC del 95%, 0.72-1.00; p = 0.05 o 0.84; IC 95%, 0.71-0.99; p = 0.03 después del ajuste de factores básales) en comparación con placebo. Atorvastatina a dosis de 80 mg disminuyó significativamente el riesgo de eventos coronarios mayores (HR 0.67; CI 95% 0.51-0.89; p = 0.006), cualquier evento de enfermedad coronaria (HR 0.60; CI del 95%, 0.48-0.74; p < 0.001) y de los procedimientos de revascularización (HR 0.57; CI del 95%, 0.44-0.74; p < 0.001). En un análisis posterior, atorvastatina a dosis de 80 mg disminuyó la incidencia de evento vascular cerebral isquémico (218/2,365, 9.2% vs. 274/2,366, 11.6%, p = 0.01) y aumentó la incidencia de evento vascular cerebral hemorrágico (55/2,365, 2.3% vs. 33/2,366, 1.4%, p = 0.02) en comparación con placebo. La incidencia de evento vascular cerebral hemorrágico mortal fue similar entre los grupos (17 con atorvastatina vs. 18 con placebo). Se demostró disminución en el riesgo de eventos cardiovasculares con atorvastatina a dosis de 80 mg en todos los grupos de pacientes, excepto quienes ingresaron al estudio con un evento vascular cerebral hemorrágico y presentaron un evento vascular hemorrágico recurrente (7 con atorvastatina vs. 2 con placebo). En pacientes tratados con atorvastatina a dosis de 80 mg hubo menos eventos vasculares cerebrales de cualquier tipo (265 con atorvastatina vs. 311 con placebo) y menos eventos de enfermedad coronaría (123 con atorvastatina vs. 204 con placebo). La mortalidad global fue similar en los grupos de tratamiento (216 con atorvastatina vs. 211 con placebo). La incidencia global de eventos adversos y eventos adversos serios fue similar entre los grupos de tratamiento. Prevención secundaria de eventos cardiovasculares: En el estudio Treating to New Targets (TNT), se valoró el efecto de atorvastatina a dosis de 80 mg/día vs. atorvastatina 10 mg/día sobre la disminución de eventos cardiovasculares en 10,001 sujetos (94% de raza blanca 81% hombres, 38% ≥ 65 años) con enfermedad coronaria clínicamente evidente que habían alcanzado una cifra blanco de C-LDL < 130 mg/dl después de concluir un periodo de ocho semanas en estudio abierto con atorvastatina 10 mg/día. Los sujetos se distribuyeron en forma aleatoria para recibir 10 mg u 80 mg/día de atorvastatina y se vigilaron durante un promedio de 4.9 años. Las cifras promedio de C-LDL, TC, TG, C no-HDL y C-HDL a las doce semanas fueron 73, 145, 128, 98 y 47 mg/dl durante el tratamiento con 80 mg de atorvastatina y 99, 177, 152, 129 y 48 mg/dl durante el tratamiento con 10 mg de atorvastatina. El tratamiento con atorvastatina a dosis de 80 mg/día disminuyó significativamente la tasa de eventos cardiovasculares mayores (MCVE, por sus siglas en inglés) (434 eventos en el grupo de 80 mg/día vs. 548 en el de 10 mg/día) con una disminución relativa del riesgo de 22%. Atorvastatina a dosis de 80 mg disminuyó significativamente el riesgo de: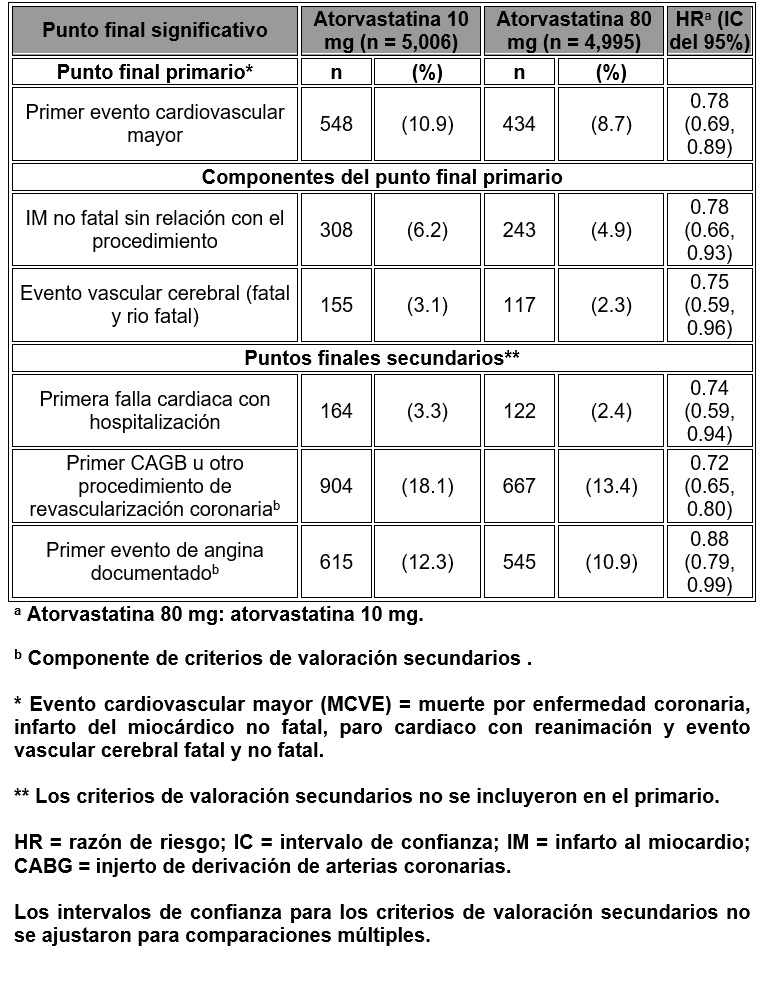
No hubo diferencia significativa entre los grupos de tratamiento para la mortalidad de todas las causas: 282 muertes (5.6%) en el grupo con 10 mg/día vs. 284 (5.7%) en el de 80 mg /día de atorvastatina. Los porcentajes de sujetos que experimentaron muerte cardiovascular, incluidos los componentes de la muerte por enfermedad coronaria y evento vascular cerebral fatal, fueron numéricamente menores en el grupo de tratamiento con 80 mg que en el de 10 mg de atorvastatina. Los porcentajes de sujetos que experimentaron una muerte de origen no cardiovascular fueron numéricamente mayores en el grupo con tratamiento de 80 mg que en el de 10 mg de atorvastatina. En el estudio Incremental Decrease in EndPoints Through Aggressive Lipid Lowering (IDEAL), se comparó el tratamiento con atorvastatina a dosis de 80 mg/día con el de simvastatina a dosis de 20-40 mg/día en 8,888 sujetos de hasta 80 años de edad con antecedente de enfermedad coronaria para valorar si podría lograrse la disminución del riesgo cardiovascular (CV). Los pacientes fueron principalmente hombres (81%) de raza blanca (99%) con una edad promedio de 61.7 años y un C-LDL promedio de 121.5 mg/dl en el momento de la distribución aleatoria; 76% estaba bajo tratamiento con estatinas. En este estudio prospectivo, aleatorizado, abierto, de punto final ciego, los sujetos fueron seguidos por un tiempo promedio de 4.8 años. Las cifras promedio de C-LDL, CT, TG, C-HDL y no-HDL en la semana 12 fueron 78, 145, 115, 45 y 100 mg/dl, en ese orden, durante el tratamiento con 80 mg de atorvastatina y 105, 179, 142, 47 y 132 mg/dl, respectivamente, durante el tratamiento con 20-40 mg de simvastatina, respectivamente. No hubo diferencia significativa entre los grupos de tratamiento para el punto final primario, la tasa del primer evento coronario mayor (enfermedad coronaria fatal, IM no fatal y reanimación después de un paro cardiaco): 411 (9.3%) en el grupo de atorvastatina a dosis de 80 mg/día vs. 463 (10.4%) en el de simvastatina a dosis de 20-40 mg/día, HR 0.89, IC del 95% (0.78, 1.01), p = 0.07. No hubo diferencias significativas entre los grupos de tratamiento en cuanto a la mortalidad por todas las causas; 366 (8.2%) en el grupo con atorvastatina a dosis de 80 mg/día vs. 374 (8.4%) en el grupo de simvastatina a dosis de 20-40 mg/día. Los porcentajes de sujetos que experimentaron muerte cardiovascular o de origen no cardiovascular, fueron similares para el grupo de atorvastatina de 80 mg y el de 20-40 mg de simvastatina. Hipercolesterolemia familiar heterocigota en pacientes pediátricos: En un estudio doble ciego, controlado con placebo, seguido por una fase de tipo abierto, 187 niños y niñas en periodo previo a la menarca de 10 a 17 años de edad (edad promedio 14.1 años) con hipercolesterolemia familiar heterocigota (FH) o hípercolesterolemia grave se distribuyeron en forma aleatoria para recibir atorvastatina (n = 140) o placebo (n = 47) durante 26 semanas. La inclusión en el estudio requería: (1) una cifra basal de C-LDL > 190 mg/dl o 2) un C-LDL basal ≥ 160 mg/dl y el antecedente familiar positivo de FH o enfermedad cardiovascular prematura demostrada en un pariente de primero o segundo grado. La cifra basal promedio de C-LDL fue de 218.6 mg/dl (variación: 138.5-385.0 mg/dl) en el grupo de atorvastatina, en comparación con 230.0 mg/dl (variación: 160.0-324.5 mg/dl) en el grupo con placebo. La dosis de atorvastatina (una vez al día) fue de 10 mg en las primeras cuatro semanas y se aumentó de manera gradual hasta 20 mg si la cifra de C-LDL era mayor de 130 mg/dl. El número de pacientes tratados con atorvastatina que requirió aumento de la dosis hasta 20 mg después de la semana 4 durante la fase doble ciego del estudio fue de 80 (57.1%). Atorvastatina disminuyó significativamente las cifras plasmáticas de C total, C-LDL, triglicéridos y apolipoproteína B durante la fase doble ciego de 26 semanas del estudio (véase tabla 1).
Tabla 1. Efectos en la disminución de lípidos con de atorvastatina en niños y niñas adolescentes con hipercolesterolemia familiar heterocigota o hipercolesterolemia grave (cambio porcentual promedio respecto de la basal en el criterio de valoración del grupo de intención a tratar)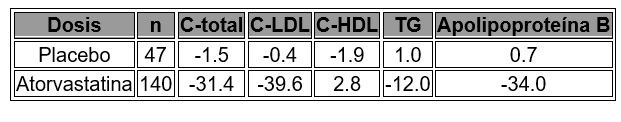
La cifra promedio de C-LDL alcanzada fue de 130.7 mg/dl (variación: 70.0-242.0 mg/dl) en el grupo de atorvastatina, en comparación con 228.5 mg/dl (variación: 152.0-385.0 mg/dl) en el grupo con placebo durante la fase doble ciego de 26 semanas. En este estudio controlado, limitado, no hubo efecto detectable sobre el crecimiento o la maduración sexual en niños o la duración del ciclo menstrual en niñas. Atorvastatina no se ha estudiado en esquemas clínicos controlados que incluyan niñas prepuberales, o bien aquellas menores de 10 años de edad. No se han estudiado la seguridad y eficacia de las dosis mayores de 20 mg en estudios controlados de niños. No se ha establecido la eficacia a largo plazo del tratamiento con atorvastatina en la infancia para disminuir la morbilidad y mortalidad en la edad adulta.
Contraindicaciones: Atorvastatina está contraindicada en pacientes que tienen: Hipersensibilidad a cualquier componente de este medicamento. Enfermedad hepática activa o elevaciones persistentes inexplicables de transaminasas séricas que excedan tres veces el límite superior de lo normal, o quienes estén: Embarazadas, lactando, o con potencial reproductivo que no estén tomando medidas anticonceptivas adecuadas. Atorvastatina debe administrarse a mujeres en edad reproductiva sólo cuando sea altamente improbable que dichas pacientes conciban y hayan sido informadas de las amenazas potenciales para el feto.
Restricciones de uso durante el embarazo y la lactancia: Atorvastatina está contraindicada en mujeres embarazadas. Las mujeres con potencial reproductivo deben usar medidas anticonceptivas adecuadas. La atorvastatina debe ser administrada a mujeres en edad reproductiva sólo cuando sea altamente improbable que dichas pacientes conciban y hayan sido informadas de las amenazas potenciales para el feto. Atorvastatina está contraindicada en mujeres que estén lactando. No se sabe si este fármaco es excretado en la leche humana. Debido a las reacciones adversas potenciales en infantes lactantes, las mujeres que tomen atorvastatina no deben lactar.
Reacciones secundarias y adversas: Atorvastatina generalmente es bien tolerada. Las reacciones adversas han sido usualmente leves y transitorias. En la base de datos de estudios clínicos de atorvastatina controlados con placebo compuesta por 16,066 (8,755 ETURION® frente a 7,311 placebo) pacientes tratados durante una mediana de 53 semanas, 5.2% de los pacientes de atorvastatina suspendieron a causa de reacciones adversas en comparación con 4.0% de los pacientes de placebo. Los efectos adversos más frecuentes (≥ 1%) asociados con la terapia de atorvastatina, en pacientes que participaron en estudios clínicos controlados fueron: Infecciones e infestaciones: Nasofaringitis. Trastornos del metabolismo y la nutrición: Hiperglucemia. Trastornos respiratorios, torácicos y mediastinales: Dolor faringo-laríngeo, epistaxis. Trastornos gastrointestinales: Diarrea, dispepsia, náuseas, flatulencia. Trastornos musculoesqueléticos y del tejido conectivo: Artralgia, dolor en las extremidades, dolor musculoesquelético, espasmos musculares, mialgia, inflamación articular. Investigaciones: Anormalidades de las pruebas de función hepática, aumento de la creatin-fosfoquinasa en sangre. Otros efectos adversos informados en estudios clínicos de atorvastatina controlados con placebo incluyen: Trastornos psiquiátricos: Pesadillas. Trastornos oculares: Visión borrosa. Trastornos del oído y el laberinto: Tinnitus. Trastornos gastrointestinales: Malestar abdominal, eructos. Trastornos hepatobiliares: Hepatitis, colestasis. Trastornos de la piel y el tejido subcutáneo: Urticaria. Trastornos musculoesqueléticos y del tejido conectivo: Fatiga muscular, dolor en el cuello. Trastornos generales y problemas del sitio de administración: Malestar general, pirexia. Investigaciones: Leucocituria. No todos los efectos mencionados antes se han asociado de forma causal con la terapia con atorvastatina. Pacientes pediátricos: Los pacientes tratados con atorvastatina tuvieron un perfil de experiencia adversa generalmente similar a aquel de los pacientes tratados con placebo; las experiencias adversas más comunes observadas en ambos grupos, sin importar la evaluación de causalidad, fueron infecciones. En la experiencia postcomerciaiización se han reportado los siguientes efectos indeseables adicionales: Trastornos sanguíneos y del sistema linfático: Trombocitopenia. Trastornos del sistema inmunológico: Reacciones alérgicas (incluyendo anafilaxia). Lesiones, envenenamiento y complicaciones del procedimiento: Ruptura de tendón. Trastornos nutricionales y del metabolismo: Aumento de peso. Trastornos del sistema nervioso: Hipoestesia, amnesia, mareo, disgeusia. Trastornos gastrointestinales: Pancreatitis. Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, eritema multiforme, erupciones bullosas. Trastornos musculoesqueléticos y del tejido conectivo: Rabdomiólisis, dolor de espalda. Trastornos generales y afecciones en el sitio de administración: Dolor torácico, edema periférico, fatiga.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis, mutagénesis, fertilidad: Atorvastatina no fue carcinogénica en ratas. La dosis máxima utilizada fue 63 veces mayor que la dosis más alta en humanos (80 mg/día) sobre una base mg/kg de peso corporal y 8 a 16 veces mayor sobre valores de ABC (0-24). En un estudio de 2 años en ratones, la incidencia de adenomas hepatocelulares en machos y carcinomas hepatocelulares en hembras se incrementó con la dosis máxima utilizada, la cual fue 250 veces mayor a la dosis más alta en humanos sobre una base mg/kg de peso corporal. La exposición sistémica fue de 6 a 11 veces mayor en AUC (0-24). Todos los demás fármacos químicamente similares de esta clase han inducido tumores tanto en ratones como en ratas a múltiplos de 12 a 125 veces las dosis clínicas más altas recomendadas sobre una base mg/kg de peso corporal. Atorvastatina no demostró potencial mutagénico ni clastogénico en cuatro pruebas in vitro con y sin activación metabólica o en un ensayo in vivo. Fue negativo en la prueba AMEs con Salmonella typhimurium y Escherichia coli, y en el ensayo de mutación precoz HGPRT in vitro en células pulmonares de hámster chino. Atorvastatina no produjo aumentos significativos en aberraciones cromosomales en el ensayo de células pulmonares de hámster Chino in vitro y fue negativo en la prueba de micronúcleo en ratón in vivo. No se observaron efectos adversos en la fertilidad o reproducción en ratas macho a las que se les administró dosis de atorvastatina de hasta 175 mg/kg/día o en ratas hembra a las que se les dio dosis de hasta 225 mg/kg/día. Estas dosis son de 100 a 140 veces la dosis humana máxima recomendada en una base mg/kg. Atorvastatina no produjo efectos adversos en los parámetros de espermas o semen, ni en la histopatología del órgano reproductivo en perros a los que se les administraron dosis de 10, 40 o 120 mg/kg durante dos años.
Interacciones medicamentosas y de otro género: El riesgo de miopatía durante el tratamiento con inhibidores de HMG-CoA reductasa se incrementa con administración concomitante de ciclosporina, derivados del ácido fíbrico, dosis modificadoras de lípidos de niacina o inhibidores del citocromo P-450 3A4 (por ejemplo, eritromicina, y antimicóticos azoles) (véase Dosis y vía de administración-Uso en combinación con otros compuestos medicinales y Precauciones generales-Efectos musculoesqueléticos). Inhibidores del citocromo P-450 3A4: Atorvastatina se metaboliza mediante citocromo P-450 3A4. La administración concomitante de atorvastatina con inhibidores del citocromo P-450 3A4 puede conducir a aumentos en las concentraciones plasmáticas de atorvastatina. El grado de interacción y potenciación de los efectos depende de la variabilidad del efecto sobre el citocromo P-450 3A4. Inhibidores transportadores: Atorvastatina y los metabolitos de la atorvastatina son sustratos del transportador OATP1B1. Los inhibidores de OATP1B1 (por ejemplo, ciclosporina) pueden incrementar la biodisponibilidad de atorvastatina. La administración concomitante de atorvastatina 10 mg y ciclosporina 5.2 mg/kg/día resultó en un aumento 7.7 veces en exposición a atorvastatina (véase Dosis y vía de administración-Uso en combinación con otros compuestos medicinales). Eritromicina/claritromicina: La coadministración de atorvastatina y eritromicina (500 mg cuatro veces al día), o claritromicina (500 mg dos veces al día) inhibidores conocidos del citocromo P-450 3A4, fue asociada con concentraciones plasmáticas más altas de atorvastatina (véase Precauciones generales-Efectos musculoesqueléticos). Inhibidores de proteasa: La coadministración de atorvastatina e inhibidores de proteasa, inhibidores conocidos de citocromo P-450 3A4, fue asociada con el aumento de concentraciones plasmáticas de atorvastatina (véase Propiedades farmacocinéticas). Clorhidrato de diltiazem: La coadministración de atorvastatina (40 mg) con diltiazem (240 mg) fue asociada con concentraciones plasmáticas mayores de atorvastatina. Cimetidina: Se realizó un estudio de interacción de atorvastatina con cimetidina, y no se observó interacción clínicamente significativa. Itraconazol: La administración concomitante de atorvastatina (20 a 40 mg) e itraconazol (200 mg) fue asociada con un incremento en el ABC de atorvastatina. Jugo de toronja: Contiene uno o más componentes que inhiben CYP 3A4 y pueden incrementar las concentraciones plasmáticas de atorvastatina, especialmente con un consumo excesivo de jugo de toronja ( > 1.2 litros por día). Inductores del citocromo P-450 3A: La administración concomitante de atorvastatina con inductores del citocromo P-450 3A4 (por ejemplo, efavirenz, rifampicina) puede conducir a reducciones variables en las concentraciones plasmáticas de atorvastatina. Debido al mecanismo de interacción dual de rifampicina, (inducción del citocromo P-450 3A4 e inhibición del transportador de captación de hepatocitos OATP1B1), se recomiendan coadministraciones simultáneas de atorvastatina con rifampicina, ya que la administración retardada de atorvastatina después de la administración de rifampicina ha sido asociada con una reducción significativa en las concentraciones plasmáticas de atorvastatina. Antiácidos: La coadministración de atorvastatina con una suspensión de antiácido oral que contiene magnesio e hidróxidos de aluminio, disminuyó las concentraciones plasmáticas de atorvastatina en aproximadamente 35%; sin embargo, no se alteró la reducción de C-LDL. Antipirina: Debido a que atorvastatina no afecta la farmacocinética de antipirina, no se esperan interacciones con otros fármacos metabolizados mediante la misma isoenzima de citocromo. Colestipol: Las concentraciones plasmáticas de atorvastatina fueron más bajas (aproximadamente 25%) cuando colestipol fue administrado con atorvastatina. Sin embargo, los efectos de lípidos fueron mayores cuando atorvastatina y colestipol fueron coadministrados que cuando cada fármaco se administró por separado. Digoxina: Las concentraciones plasmáticas en estado estable de digoxina no se vieron afectadas cuando se coadministraron dosis múltiples de digoxina y 10 mg de atorvastatina. Sin embargo, las concentraciones de digoxina aumentaron aproximadamente 20% tras administración de digoxina con 80 mg de atorvastatina diariamente. Los pacientes que toman digoxina deben ser monitoreados de manera apropiada. Azitromicina: La coadministración de atorvastatina (10 mg una vez al día) y azitromicina (500 mg una vez al día) no alteró las concentraciones plasmáticas de atorvastatina. Anticonceptivos orales: La coadministración con un anticonceptivo oral que contiene noretindrona y etinil estradiol aumentó los valores ABC de noretindrona y etinil estradiol por aproximadamente 30% y 20%. Estos aumentos deben considerarse cuando se selecciona un anticonceptivo oral para una mujer que toma atorvastatina. Warfarina: Se realizó un estudio de interacción de Atorvastatina con warfarina, y no se observaron inter