FACTIVE®-5
PFIZER
Denominación genérica: Gemifloxacino
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: Mesilato de gemifloxacino equivalente a 320mg de gemifloxacino. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: FACTIVE®-5 está indicado en el tratamiento de infecciones causadas por cepas sensibles de los microorganismos designados, en las afecciones enumeradas a continuación. Ver dosis y vía de administración. Exacerbación bacteriana de bronquitis crónica causada por Streptococcus pneumoniae, Haemophilus influenzae, Haemophilus parainfluenzae, o Moraxella catarrhalis. Sinusitis bacteriana aguda causada por Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus (sólo las cepas sensibles a la meticilina), Klebsiella pneumoniae o Escherichia coli. Neumonía adquirida en la comunidad (de gravedad leve a moderada) causada por Streptococcus pneumoniae (incluso cepas resistentes a múltiples medicamentos [SPMR])*, Haemophilus influenzae, Moraxella catarrhalis, Mycoplasma pneumoniae, Chlamydophila pneumoniae, o Klebsiella pneumoniae**. *El Streptococcus pneumoniae resistente a múltiples medicamentos incluye las cepas anteriormente conocidas como SPPR (Streptococcus pneumoniae resistente a la penicilina) y son cepas resistentes a dos o más de los siguientes antibióticos: penicilina, cefalosporinas de segunda generación (por ejemplo, cefuroxima), macrólidos, tetraciclinas y trimetoprim/sulfametoxazol. Infecciones de vías urinarias no complicadas. (E. coli, K. pneumoniae, P. mirabilis, Staphylococcus saporophyticus). Para reducir el desarrollo de bacterias resistentes a fármacos y mantener la eficacia de FACTIVE®-5 y de otros fármacos antibacterianos, FACTIVE®-5 sólo debe usarse para tratar infecciones de las que se sabe o se sospecha fehacientemente que son causadas por bacterias sensibles. En aquellos casos en los que se dispone de cultivos e información sobre la sensibilidad, se deberán considerar estos datos en la selección o la modificación del tratamiento antibacteriano. En ausencia de esos datos, los patrones de sensibilidad y epidemiología locales pueden contribuir a la selección empírica de la terapia.
Farmacocinética y farmacodinamia: Farmacocinética: la farmacocinética del gemifloxacino es prácticamente lineal en un rango de dosis de 40 mg a 640 mg. Se observó una acumulación mínima de gemifloxacino luego de dosis orales múltiples de hasta 640 mg por día, durante 7 días (acumulación media < 20%). Luego de la administración de dosis orales repetidas de 320 mg de gemifloxacino, una vez por día, el estado de equilibrio se alcanza al tercer día de tratamiento. Absorción y biodisponibilidad: El gemifloxacino, administrado como comprimido oral, se absorbe rápidamente en el tracto gastrointestinal. Luego de la administración del comprimido por vía oral, se observaron concentraciones máximas de gemifloxacino en el plasma entre la 0,5 hora y las 2 horas siguientes, y la biodisponibilidad absoluta del comprimido de 320 mg fue, en promedio, de alrededor del 71% (IC 95%: 60%-84%). Luego de dosis orales repetidas de 320 mg en sujetos sanos, la media ± DE de las concentraciones plasmáticas máximas de gemifloxacino (Cmáx) y la exposición sistemática al fármaco (ABC (0-24)) fueron 1,61 ± 0,51 mg/ml (rango 0,70-2,62 (mg/ml) y 9,93 ± 3,07 mg•h/ml (rango 4,71-20,1 mg•h/ml), respectivamente. En los pacientes con infecciones del tracto urinario y respiratorio (n=1423), se calcularon estimaciones similares de exposición sistémica al fármaco usando un análisis farmacocinético poblacional (ABC media geométrica (0-24), 8,36 mg•h/ml; rango 3,2 - 47,7 mg•h/ml). La farmacocinética de gemifloxacino no resultó significativamente alterada cuando se administró una dosis de 320 mg con una comida de alto contenido graso. Por lo tanto, los comprimidos de FACTIVE®-5 pueden administrarse independientemente de las comidas. Distribución: la unión in vitro del gemifloxacino a las proteínas plasmáticas en sujetos sanos es de alrededor del 60 al 70%, y es independiente de la concentración. Luego de dosis repetidas, la unión a las proteínas plasmáticas in vivo en sujetos sanos ancianos y jóvenes osciló entre un 55% y un 73%, y la edad no influyó en absoluto. La insuficiencia renal no afecta significativamente la unión del gemifloxacino a las proteínas. La relación concentración del gemifloxacino en sangre/plasma fue 1,2:1. La media geométrica para Vdss/F es de 4,18 L/kg (con un rango de 1,66 - 12,12 l/kg). Luego de su administración oral, el gemifloxacino se distribuye extensamente por todo el organismo. Las concentraciones de gemifloxacino en líquido de lavado broncoalveolar exceden a las del plasma. El gemifloxacino penetra perfectamente en los líquidos y tejido pulmonares. Luego de cinco dosis diarias de 320 mg de gemifloxacino, las concentraciones en plasma, macrófagos broncoalveolares, líquido de la capa epitelial y la mucosa bronquial fueron, a las 2 horas aproximadamente, las incluidas en la Tabla 1: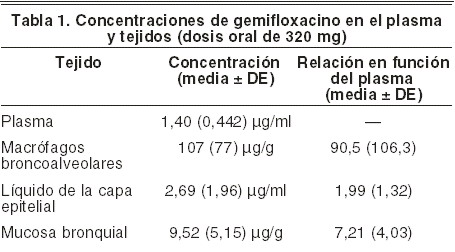
Metabolismo: el gemifloxacino es metabolizado por el hígado en un grado limitado. El compuesto inalterado es el componente predominante, relacionado al fármaco, detectado en el plasma (aproximadamente 65%) hasta 4 horas después de la administración de la dosis. Todos los metabolitos que se forman son de escasa importancia ( < 10% de la dosis oral administrada); los principales son: N-acetil gemifloxacino, el E-isómero de gemifloxacino y el glucurónido carbamil del gemifloxacino. Las enzimas del citocromo P450 no desempeñan una función fundamental en el metabolismo del gemifloxacino, y éste no inhibe la actividad metabólica de estas enzimas en forma significativa. Excreción: el gemifloxacino y sus metabolitos se excretan a través de dos vías de excreción. Luego de una administración oral de gemifloxacino a sujetos sanos, una media (± DE) de 61 ± 9,5% de la dosis se excretó en las heces y 36 ± 9,3% en la orina como fármaco inalterado y sus metabolitos. La depuración renal media (± DE) luego de dosis repetidas de 320 mg fue de aproximadamente 11,6 ± 3,9 l/h (rango 4,6-17,6 l/h), lo que indica que en la excreción renal del gemifloxacino participa la secreción activa. La media (± DE) de la vida media de eliminación plasmática en estado de equilibrio luego de administrar 320 mg a sujetos sanos fue de alrededor de 7 ± 2 horas (rango 4-12 horas). Poblaciones especiales: niños: no se ha estudiado la farmacocinética del gemifloxacino en niños. Ancianos: en los sujetos adultos, la edad no afecta la farmacocinética del gemifloxacino. Sexo: no hay diferencias importantes entre la farmacocinética del gemifloxacino en hombres y mujeres cuando se tienen en cuenta las diferencias en el peso corporal. Los estudios farmacocinéticos poblacionales indican que luego de la administración de 320 mg de gemifloxacino, los valores del ABC fueron cerca de un 10% superior en las mujeres sanas que en los hombres sanos. Los hombres y las mujeres presentaron ABC medias de 7,98 mg•h/ml (rango 3,21 - 42,71 mg•h/ml) y 8,80 mg•h/ml (rango 3,33 - 47,73 mg•h/ml), respectivamente. No es necesario ningún ajuste de dosis en base al sexo. Insuficiencia hepática: se estudió la farmacocinética de una dosis única de 320 mg de gemifloxacino en pacientes con enfermedad hepática leve (Clase A según la escala de Child-Pugh) a moderada (Clase B según la escala de Child-Pugh). En estos pacientes con disfunción hepática se observó un aumento medio en el ABC (0-inf) del 34% y un aumento medio en la Cmáx del 25% en comparación con los voluntarios sanos. También se estudió la farmacocinética de una dosis única de 320 mg de gemifloxacino en pacientes con disfunción hepática grave (Clase C de la escala Child-Pugh). En estos sujetos con disfunción hepática hubo un aumento medio en el ABC (0-inf) del 45% y un aumento medio en la Cmáx de 41% en comparación con los voluntarios sanos. Se considera que el promedio de estos aumentos farmacocinéticos carecen de relevancia clínica. No hubo cambios importantes en la vida media de eliminación plasmática en los pacientes con insuficiencia hepática leve, moderada o grave. No se recomienda ajustar la dosis en los pacientes con insuficiencia hepática leve (clase A de la escala Child-Pugh), moderada (clase B de la escala Child-Pugh) o grave (clase C de la escala Child-Pugh) (consultar dosis y vía de administración). Insuficiencia renal: en los pacientes con insuficiencia renal, los resultados de los estudios de farmacocinética poblacional y farmacología clínica con dosis repetidas de 320 mg indican que la depuración de gemifloxacino se reduce y la eliminación plasmática se prolonga, lo que conduce a un aumento de alrededor del 70% en los valores del ABC. En los estudios farmacocinéticos, la Cmáx de gemifloxacino no resultó significativamente alterada en los sujetos con insuficiencia renal. En los pacientes con una depuración de la creatinina > 40 ml/min no es necesario ajustar la dosis. Se recomienda modificar la posología en los pacientes cuya depuración de creatinina es ≤40 ml/min (ver dosis y vía de administración). La hemodiálisis elimina del plasma alrededor del 20% al 30% de una dosis oral de gemifloxacino. Fotosensibilidad potencial: en un estudio de respuesta cutánea a radiación visible y ultravioleta realizado en 40 voluntarios sanos, se determinó la dosis eritematosa mínima (DEM) luego de la administración de 160 mg de gemifloxacino una vez al día o 320 mg de gemifloxacino una vez al día o 500 mg de ciprofloxacino dos veces por día o un placebo, durante 7 días. En 5 de las 6 longitudes de onda analizadas (295-430 nm), el potencial de fotosensibilidad del gemifloxacino no difirió del placebo lo suficiente como para alcanzar significancia estadística. A 365 nm (región UVA), el gemifloxacino mostró un potencial de fotosensibilidad similar al de ciprofloxacino 500 mg dos veces al día, y el potencial de fotosensibilidad de ambos fármacos fue estadísticamente superior al del placebo. Rara vez se informaron reacciones de fotosensibilidad en los estudios clínicos con gemifloxacino (0,039%) (consultar reacciones secundarias y adversas).
Contraindicaciones: El gemifloxacino está contraindicado en pacientes con antecedentes de hipersensibilidad a este medicamento, a las fluoroquinolonas o a cualquiera de los componentes de la fórmula. Advertencias: no se ha determinado la seguridad y la eficacia de FACTIVE®-5 en niños, adolescentes (menores de 18 años de edad), mujeres embarazadas y durante la lactancia (consultar Precauciones generales: sub-secciones de Uso pediátrico, Embarazo y lactancia.)
Precauciones generales: General: si se prescribe FACTIVE®-5 sin que exista una infección bacteriana comprobada o una fuerte sospecha de infección bacteriana, es improbable que el paciente resulte beneficiado, y aumenta el riesgo de que desarrolle bacterias resistentes al fármaco. Erupción cutánea (rash): en estudios clínicos sobre el tratamiento con gemifloxacino durante 5 días, el índice general de erupción cutánea (rash) relacionada con el fármaco fue de 1,1%, similar al índice de los comparadores (0,7%). Aproximadamente 0,1% de todos los pacientes que recibieron gemifloxacino durante 5 días describieron la erupción cutánea como de intensidad grave. La duración del tratamiento con gemifloxacino más allá de los 5 días hace que la incidencia de la erupción cutánea aumente. Debe descontinuarse el tratamiento con gemifloxacino en los pacientes que desarrollan una erupción cutánea (rash) mientras están en tratamiento (consultar Reacciones secundarias y adversas). En los estudios clínicos con FACTIVE®-5 muy rara vez se informaron reacciones de fotosensibilidad (ver Farmacocinética y farmacodinamia en humanos.) Sin embargo, como con todos los fármacos de su clase, se recomienda que los pacientes eviten la exposición innecesaria a la luz solar potente o a los rayos UV artificiales (por ejemplo, lámpara solar o en solarium), y se le debe recomendar el uso adecuado de protectores solares de amplio espectro si están expuestos a la luz solar brillante. Si se sospecha de una reacción de fotosensibilidad, se deberá descontinuar el tratamiento. Efectos renales: es necesario modificar el régimen posológico en los pacientes con insuficiencia renal (depuración de creatinina ≤40 ml/min) (ver dosis y vía de administración). Se debe mantener una adecuada hidratación de los pacientes que reciben gemifloxacino para prevenir la formación de orina concentrada. Efectos sobre el intervalo QT: en algunos pacientes, las fluoroquinolonas pueden prolongar el intervalo QT. Se debe evitar el gemifloxacino en pacientes con antecedentes de prolongación en el intervalo QTc, en pacientes con trastornos electrolíticos (hipopotasemia o hipomagnesemia) y en los pacientes que toman agentes antiarrítmicos clase IA (por ejemplo, quinidina, procainamida) o clase III (por ejemplo, amiodarona, sotalol). No se realizaron estudios farmacocinéticos entre el gemifloxacino y fármacos que prolongan el intervalo QTc como la eritromicina, los antipsicóticos y los antidepresivos tricíclicos. El gemifloxacino debe usarse con precaución cuando se administra concomitantemente con estos fármacos, así como en pacientes con afecciones proarrítmicas, como la bradicardia clínicamente significativa o la isquemia aguda de miocardio. No se produjo morbilidad o mortalidad cardiovascular atribuible a la prolongación del intervalo QTc en más de 8119 pacientes tratados con gemifloxacino, incluyendo 707 pacientes que recibían concomitantemente fármacos conocidos por prolongar el intervalo QTc y 7 pacientes con hipopotasemia. La probabilidad de la prolongación del intervalo QTc puede aumentar al incrementar la dosis del fármaco; por lo tanto, no debe excederse la dosis recomendada, especialmente en aquellos pacientes con disfunción hepática o renal en los que la Cmáx y el ABC son ligeramente más altos. La prolongación del intervalo QTc puede producir un aumento en el riesgo de arritmias ventriculares incluyendo torsades de pointes. El cambio máximo en el intervalo QTc se produce luego de aproximadamente 5-10 horas posteriores a la administración oral del gemifloxacino. Reacciones de hipersensibilidad: se informaron casos de hipersensibilidad fatal ocasional y/o reacciones anafilácticas en pacientes que recibieron tratamiento con fluoroquinolonas. Se han presentado informes de reacciones anafilácticas en pacientes tratados con gemifloxacino. Algunas de las reacciones de hipersensibilidad informadas en pacientes que recibían tratamiento con fluoroquinolonas se produjeron luego de la primera dosis. Algunas reacciones estuvieron acompañadas de colapso cardiovascular, hipotensión/choque, convulsiones, pérdida del conocimiento, hormigueo, angioedema (incluyendo edema/inflamación de la lengua, laríngeo, de garganta o facial), obstrucción de las vías aéreas (incluyendo broncoespasmo, respiración entrecortada e insuficiencia respiratoria aguda), disnea, urticaria, prurito y otras reacciones cutáneas graves. *Se debe suspender de inmediato el uso de la gemifloxacino ante la aparición de cualquier signo de erupción cutánea por hipersensibilidad de tipo I inmediata, o cualquier otra manifestación de una reacción de hipersensibilidad; se deberá evaluar si es necesario continuar el tratamiento con fluoroquinolonas. Como con otros fármacos, las reacciones agudas de hipersensibilidad graves pueden requerir un tratamiento con epinefrina u otras medidas de resucitación, incluyendo los aportes de oxígeno, líquidos por vía intravenosa, antihistamínicos, corticoides, aminas presoras y la permeabilidad de las vías aéreas según se indique clínicamente (ver Reacciones secundarias y adversas). En pacientes tratados con fluoroquinolonas se comunicaron eventos graves, y ocasionalmente fatales, algunos debidos a la hipersensibilidad y algunos de etiología incierta. Estos eventos pueden ser graves y generalmente se producen luego de la administración de dosis múltiples. Las manifestaciones clínicas usualmente incluyen un nuevo episodio de fiebre, y una o más de las siguientes: erupción cutánea o reacciones dermatológicas graves (por ejemplo, necrólisis epidérmica tóxica, síndrome de Stevens-Johnson), vasculitis, artralgia, mialgia, enfermedad del suero, neumonitis alérgica, nefritis intersticial, insuficiencia o falla renal aguda, hepatitis, ictericia, necrosis o insuficiencia hepática aguda, anemia, incluyendo la hemolítica y la aplásica, trombocitopenia, incluyendo la púrpura trombocitopénica trombótica, leucopenia; agranulocitosis, pancitopenia; y/o otras anomalías hematológicas. Neuropatía periférica: en pacientes tratados con quinolonas se ha informado de casos raros de polineuropatía axonal sensitiva o sensitivomotora que afectaba axones pequeños o grandes provocando parestesia, hipoestesias, disestesias y debilidad. Efectos en los tendones: en los pacientes tratados con quinolonas se informaron roturas de los tendones del hombro, de la mano o del tendón de Aquiles u otros tendones que exigen reparación quirúrgica o que producen una discapacidad prolongada. Los informes de vigilancia postcomercialización indican que este riesgo puede aumentar en pacientes que reciben corticosteroides en forma concomitante, especialmente en los ancianos. Se debe suspender el tratamiento con gemifloxacino si el paciente experimenta dolor, inflamación o rotura de un tendón. Los pacientes deben descansar y evitar realizar ejercicios hasta que el diagnóstico de tendinitis o rotura de tendón se haya descartado. La rotura de tendón puede producirse durante o después del tratamiento con quinolonas. Efectos sobre el SNC: en los estudios clínicos con gemifloxacino, rara vez se informaron efectos sobre el sistema nervioso central (SNC). Como con otras fluoroquinolonas, el gemifloxacino debe usarse con precaución en pacientes con enfermedades del SNC como epilepsia, o en los pacientes propensos a sufrir crisis convulsivas. Aunque no se observaron en los estudios clínicos de gemifloxacino, se informaron aumentos en la presión intracraneal y psicosis tóxica en los pacientes tratados con otras fluoroquinolonas. La estimulación del SNC que puede conducir a temblores, agitación, ansiedad, aturdimiento, confusión, alucinaciones, paranoia, depresión, insomnio y, ocasionalmente, pensamientos o actos suicidas, también puede ser causada por otras fluoroquinolonas. Si los pacientes que reciben gemifloxacino presentan alguna de estas reacciones, se deberá suspender el fármaco e instaurarse las medidas apropiadas. Colitis asociada a antibióticos: se comunicaron casos de colitis pseudomembranosa con casi todos los agentes antibacterianos, incluso con el gemifloxacino, y su gravedad puede variar de leve a grave con riesgo de muerte. Por lo tanto, es importante considerar este diagnóstico en pacientes que presentan diarrea luego de la administración de cualquier agente antibacteriano. El tratamiento con agentes antibacterianos altera la flora normal del colon y puede permitir una proliferación de clostridios. Los estudios indican que una toxina producida por el Clostridium difficile es la principal causa de la ·colitis asociada a antibióticos. Luego de confirmarse el diagnóstico de colitis pseudomembranosa, deben instaurarse medidas terapéuticas. En general, los casos leves de colitis pseudomembranosa se resuelven con sólo descontinuar el fármaco. En casos moderados a graves, se debe considerar la reposición de líquidos y electrolitos, los suplementos de proteínas y el tratamiento con fármacos antibacterianos clínicamente eficaces contra la colitis por Clostridium difficile (ver Reacciones secundarias y adversas). Información para los pacientes: a los pacientes se les debe advertir: *que sólo usen los fármacos antibacterianos, tales como FACTIVE®-5, para tratar infecciones bacterianas. No deben tratar infecciones virales (como, por ejemplo, el resfrío común). Cuando FACTIVE®-5 se receta para tratar una infección bacteriana, se les debe indicar a los pacientes que sigan estrictamente las instrucciones, a pesar de que con frecuencia se sentirán mejor al poco tiempo de iniciar el tratamiento. Pasar dosis por alto o no completar el curso completo del tratamiento puede: (1) reducir la eficacia del tratamiento inmediato y (2) aumentar la probabilidad de que la bacteria desarrolle resistencia y de que en el futuro no pueda ser tratada con FACTIVE®-5 u otro agente antibacteriano de la misma clase. *Que el uso de FACTIVE®-5 se asoció con erupciones cutáneas. Si los pacientes desarrollan una erupción cutánea deberán interrumpir el tratamiento y llamar a su médico. *Que FACTIVE®-5 puede asociarse a reacciones de hipersensibilidad, incluso reacciones anafilácticas, aun luego de una dosis única; si se presentan signos de erupción cutánea (rash) o reacción alérgica los pacientes deben interrumpir el tratamiento y procurar asistencia médica de inmediato. *Que FACTIVE®-5 puede provocar cambios en el electrocardiograma (prolongación del intervalo QTc). *Que se debe evitar el uso de FACTIVE®-5 en los pacientes que reciben agentes antiarrítmicos de clase IA (por ejemplo, quinidina, procainamida) o clase III (por ejemplo, amiodarona, sotalol). *Que FACTIVE®-5 debe usarse con precaución en pacientes que reciben fármacos que afectan el intervalo QTc como cisaprida, eritromicina, antipsicóticos y antidepresivos tricíclicos. *Que deben informar a su médico de cualquier antecedente personal o familiar de prolongación del intervalo QTc o afecciones proarrítmicas como hipopotasemia, bradicardia o isquemia de miocardio reciente. *Que deben informar a su médico sobre cualquier otro medicamento que estén recibiendo concomitantemente con FACTIVE®-5, incluso los de venta libre y los suplementos dietéticos. *Que se comuniquen con su médico si experimentan palpitaciones o síncopes durante el tratamiento con FACTIVE®-5. *Que FACTIVE®-5 puede administrarse con o sin alimentos. *Que deben consumir líquido en abundancia. *Que dentro de las 3 horas previas o las 2 horas posteriores de haber recibido FACTIVE®-5 comprimidos, no tomen antiácidos que contengan magnesio y/o aluminio, o productos que contengan sulfato ferroso (hierro), preparaciones con multivitaminas que contengan zinc u otros cationes de metales, o comprimidos masticables/disolubles de Videx® (didanosina), o el polvo para solución oral de uso pediátrico. *Que FACTIVE®-5 debe ingerirse al menos dos horas antes del sucralfato. *Que se informó fototoxicidad con el uso de ciertas quinolonas. En los estudios clínicos, el potencial de FACTIVE®-5 para causar fototoxicidad fue bajo (6/9003) cuando se administró a las dosis recomendadas. Con el propósito de cumplir con las buenas prácticas clínicas, evite la exposición excesiva a la luz solar o a la luz ultravioleta artificial (por ejemplo, camas solares). Si se presenta una reacción como una quemadura solar o una erupción cutánea, comuníquese con su médico. (ver Farmacocinética y farmacodinamia en humanos). *Que FACTIVE®-5 puede producir mareos, en cuyo caso los pacientes no deben conducir automóviles u operar maquinarias, ni realizar actividades que exijan un cierto grado de alerta mental o coordinación. *Que deben interrumpir el tratamiento con FACTIVE®-5 e informar a su médico si sienten dolor, molestias o rotura de un tendón. Los pacientes deben descansar y evitar realizar ejercicios hasta que el diagnóstico de tendinitis o rotura de tendón se haya descartado. *Que se informaron convulsiones en pacientes tratados con quinolonas, y que deben avisar a su médico si tienen antecedentes de este tipo de episodios antes de iniciar el tratamiento. *Que se observaron aumentos del Indice Normalizado Internacional (INR) o del tiempo de protrombina (TP) y/o episodios hemorrágicos cuando se administraron en forma conjunta warfarina o sus derivados, y gemifloxacino. Los pacientes deben comunicarle al médico si toman warfarina o sus derivados. Interacciones farmacológicas: en sujetos sanos, la administración de dosis repetidas de FACTIVE®-5 no afectó la farmacocinética de dosis repetidas de teofilina, digoxina o de un anticonceptivo oral con etinilestradiol/levonorgestrel. (ver Farmacocinética y farmacodinamia en humanos). La administración concomitante de FACTIVE®-5 y carbonato de calcio, cimetidina, omeprazol, o un anticonceptivo oral con estrógeno/progesterona produjo cambios menores en la farmacocinética del gemifloxacino, los que se consideraron carentes de importancia clínica (ver Farmacocinética y farmacodinamia en humanos). La administración concomitante de FACTIVE®-5 con probenecid produjo un aumento del 45% en la exposición sistémica al gemifloxacino (ver Farmacocinética y Farmacodinamia en Humanos). FACTIVE®-5 no tiene un efecto importante en la acción anticoagulante de la warfarina en sujetos sanos que están en tratamiento estable con ese fármaco. Se presentaron informes sobre aumentos en el INR y en el tiempo de protrombina (TP) con el uso de quinolonas, incluso gemifloxacino, y warfarina o sus derivados. Además, la enfermedad infecciosa y su respectivo proceso inflamatorio, la edad y el estado general del paciente son factores de riesgo para el aumento de la actividad anticoagulante. Por lo tanto, el TP, el INR y las pruebas de coagulación apropiadas deben controlarse cuidadosamente si se administra una quinolona concomitantemente con warfarina o sus derivados.
Restricciones de uso durante el embarazo y la lactancia: No se ha determinado la seguridad y la eficacia de FACTIVE®-5 en niños, adolescentes (menores de 18 años de edad), mujeres embarazadas y durante la lactancia (ver Precauciones generales).
Reacciones secundarias y adversas: Experiencia con tratamiento de 5 días: en los estudios clínicos, 3696 pacientes recibieron dosis orales diarias de 320 mg de gemifloxacino durante 5 días. La mayoría de las reacciones adversas experimentadas por los pacientes en estudios clínicos se consideraron de gravedad leve a moderada. En el 0,9% de los pacientes, el tratamiento con gemifloxacino durante 5 días se interrumpió debido a un evento adverso (posible o probablemente relacionado), principalmente a causa de diarrea (0,3%). Los antibióticos comparadores se suspendieron debido a un evento adverso a una tasa global comparable de 2,3%, principalmente por diarrea (0,5%). Los eventos adversos relacionados con el fármaco, clasificados como posible o probablemente relacionados con una frecuencia ≥1% en pacientes que recibieron 320 mg de gemifloxacino durante 5 días de tratamiento vs. el fármaco comparador (antibióticos betalactámicos, macrólidos u otras fluoroquinolonas) fueron los siguientes: diarrea 3,7% vs. 5,2%; náusea 2,4% vs. 2,8%; erupción cutánea 1,1% vs. 0,7%; disgeusia 0,4% vs. 2,4%; mareos 0,7% vs. 1,7%; dolor de cabeza 0,8% vs. 1,3%; dolor abdominal 0,8% vs. 1,1% y vómitos 0,7% vs. 1,1%. Experiencia con tratamiento de variada duración: en estudios clínicos de 8119 pacientes que recibieron dosis orales diarias de 320 mg de gemifloxacino durante periodos de 3 días a 14 días (incluso los 3696 pacientes que recibieron 5 días de tratamiento), los eventos adversos relacionados con el fármaco (posible o probablemente relacionados) con una frecuencia > 1% fueron similares en tipo y frecuencia a aquellos eventos reportados con el tratamiento por 5 días, con la excepción de la erupción cutánea (rash), la cual fue más elevada debido a la inclusión de pacientes que recibieron un tratamiento de más de 5 días de duración. El gemifloxacino parece tener un bajo potencial para producir fotosensibilidad. En los estudios clínicos, la fotosensibilidad relacionada con el tratamiento sólo se observó en el 0,067% (6/9003) de los pacientes. Los eventos adversos adicionales relacionados con el fármaco (posible o probablemente relacionados) observados en los 8119 pacientes, con una frecuencia de > 0,1% a < 1% incluyeron: dolor abdominal, anorexia, constipación, dermatitis, mareos, boca seca, dispepsia, fatiga, flatulencia, infecciones fúngicas, gastritis, moniliasis genital, prurito genital, hiperglucemia, fosfatasa alcalina aumentada, TGP aumentada, TGO aumentada, creatina fosfoquinasa aumentada, insomnio, leucopenia, prurito, somnolencia, disgeusia, trombocitopenia, urticaria, vaginitis y vómitos. Otros eventos adversos, informados en los estudios clínicos, que tienen importancia clínica potencial y que se consideraron que podrían tener una relación con el fármaco y que se presentaron en ≤0,1% de los pacientes fueron: orina anormal, visión anormal, anemia, artralgia, astenia, dolor de espalda, bilirrubinemia, disnea, eczema, eosinofília, rubor, gastroenteritis, granulocitopenia, sofocos, aumento de la GGT, aumento del nitrógeno no proteico, calambres en las piernas, moniliasis, mialgia, nerviosismo, trastornos gastrointestinales no especificados, dolor, faringitis, neumonía, trombocitopenia, temblores y vértigo (ver Precauciones). Reacciones adversas posteriores a la comercialización: a continuación se citan las reacciones adversas adicionales informadas durante el uso de FACTIVE®-5 posterior a la comercialización. Como estas reacciones se informaron voluntariamente por parte de una población de tamaño incierto, es imposible calcular fehacientemente la frecuencia de las mismas o establecer una relación de causa con la exposición a FACTIVE®-5: reacción anafiláctica, eritema multiforme, inflamación facial, hemorragia, índice normalizada internacional (INR) aumentado, intervalo QT prolongado, pirexia, insuficiencia renal, hemorragia retiniana, exfoliación cutánea, taquicardia supraventricular, síncope, accidente isquémico transitorio.
Interacciones medicamentosas y de otro género: Antiácidos / cationes divalentes y trivalentes: la disponibilidad sistémica del gemifloxacino se reduce significativamente cuando se administra concomitantemente un antiácido que contenga aluminio y magnesio (el ABC se redujo un 85%; la Cmáx se redujo un 87%). La administración de un antiácido con aluminio y magnesio, o de sulfato ferroso (325 mg) 3 horas antes o 2 horas después de la administración de gemifloxacino no alteró significativamente su disponibilidad sistémica. Por lo tanto, los antiácidos que contienen aluminio y/o magnesio, sulfato ferroso (hierro), las preparaciones multivitamínicas que contienen zinc u otros cationes de metales, o comprimidos masticables/disolubles Videx® (didanosina), o el polvo para solución oral de uso en pediatría, no deben administrarse dentro de las 3 horas previas o las 2 horas posteriores a la administración de FACTIVE®-5 comprimidos. El carbonato de calcio (1000 mg) consumido 2 horas antes o 2 horas después de la administración de gemifloxacino no mostró notable reducción de la disponibilidad sistémica del gemifloxacino. El carbonato de calcio administrado simultáneamente con el gemifloxacino produjo una pequeña reducción, clínicamente insignificante, en la exposición del gemifloxacino [el ABC (0-inf) se redujo 21% y la Cmáx disminuyó]. Sucralfato: cuando se administró sucralfato (2 g) 3 horas antes del gemifloxacino, la biodisponibilidad oral del gemifloxacino se redujo significativamente (reducción del 53% en el ABC; reducción del 69% en la Cmáx). Cuando se administró sucralfato (2 g) 2 horas después del gemifloxacino, la biodisponibilidad oral del gemifloxacino no sufrió un cambio significativo, por lo que FACTIVE®-5 debe administrarse al menos 2 horas antes del sucralfato. (Precauciones generales). Metabolismo in vitro: los resultados de los estudios de inhibición in vitro indican que la enzima (CYP450) del citocromo hepático P450 no desempeña una función importante en el metabolismo del gemifloxacino. Por lo tanto, el gemifloxacino no debería causar interacciones farmacocinéticas importantes in vivo con otros fármacos que son metabolizados por las enzimas CYP450. Teofilina: el gemifloxacino de 320 mg en estado de equilibrio no afectó la farmacocinética de dosis repetidas de teofilina (de 300 a 400 mg dos veces por día, en sujetos del sexo masculino sanos). Digoxina: la dosis de 320 mg de gemifloxacino en estado de equilibrio no afectó la farmacocinética de dosis repetidas de digoxina (0,25 mg una vez por día en sujetos del sexo masculino sanos). Anticonceptivos orales: el efecto de un producto anticonceptivo oral con estrógeno/progesterona (una vez por día, durante 21 días) en la farmacocinética del gemifloxacino (320 mg, una vez por día, durante 6 días) en mujeres sanas indica que la administración concomitante produce una reducción promedio en el ABC y la Cmáx de 19% y 12%, respectivamente. Estos cambios no se consideran clínicamente relevantes. El gemifloxacino de 320 mg en estado de equilibrio no afectó la farmacocinética de dosis repetidas de un producto anticonceptivo oral con etinilestradiol/levonorgestrel (30 mg/150 mg una vez al día durante 21 días, en mujeres sanas). Cimetidina: la coadministración de una dosis única de 320 mg de gemifloxacino con 400 mg de cimetidina, cuatro veces por día durante 7 días, produjo aumentos leves del promedio en el ABC (0-inf) y la Cmáx del gemifloxacino del 10% y 6%, respectivamente. Estos aumentos no se consideran clínicamente relevantes. Omeprazol: la coadministración de una dosis única de 320 mg de gemifloxacino con 40 mg de omeprazol una vez por día, durante 4 días, produjo aumentos leves del promedio en el ABC (0-inf) y la Cmáx del gemifloxacino del 10% y 11%, respectivamente. Estos aumentos no se consideran clínicamente relevantes. Warfarina: la administración de varias dosis de gemifloxacino (320 mg, una vez por día, durante 7 días) a sujetos sanos que estaban recibiendo un tratamiento estable con warfarina no provocó un efecto significativo en la actividad anticoagulante inducida por esta última (es decir, la relación normalizada internacional (INR) para el tiempo de protrombina) (ver Precauciones generales). Probenecid: la administración de una dosis única de 320 mg de gemifloxacino a sujetos sanos que también recibían dosis repetidas de probenecid (dosis total = 4,5 g) redujo la depuración renal media del gemifloxacino en alrededor de un 50%, lo que resultó en un aumento promedio del 45% en el ABC (0-inf) de gemifloxacino y una prolongación de la vida media promedio de 1,6 horas. La Cmáx media del gemifloxacino aumentó un 8%. Uso pediátrico: no se determinó la seguridad y la eficacia en niños y adolescentes menores de 18 años de edad. Las fluoroquinolonas, incluida el gemifloxacino, causan artropatía y osteocondrosis en animales inmaduros (ver Precauciones generales). Uso geriátrico: de la cantidad total de sujetos en estudios clínicos de gemifloxacino, el 29% (2314) tenían al menos 65 años de edad, mientras que el 11% (865) tenían al menos 75 años. No se observaron diferencias globales en la eficacia entre estos sujetos y los sujetos más jóvenes; el índice de eventos adversos para este grupo fue similar o menor que para los sujetos más jóvenes con la excepción de la incidencia de erupción cutánea que fue menor en los ancianos comparada con los pacientes menores de 40 años de edad.
Alteraciones en los resultados de pruebas de laboratorio: Cambios en las pruebas de laboratorio: + los porcentajes de los 3.696 pacientes que recibieron 320 mg de gemifloxacino durante 5 días de tratamiento y presentaron resultados anormales de las pruebas de laboratorio se citan a continuación; sin embargo, se desconoce si esas anormalidades estaban relacionadas al gemifloxacino o a una enfermedad subyacente. Bioquímica clínica: TGP aumentada (1,3%), TGO aumentada (0,9%), fosfatasa alcalina aumentada (0,2%), creatinina sérica aumentada (0,2%), creatina fosfoquinasa aumentada (0,7%), calcio disminuido (0,1%), potasio aumentado (0,7%), sodio aumentado (0,4%), proteínas totales disminuidas (0,1%), albúmina disminuida (0,2%) y bilirrubina total aumentada (0,1%). Hematología: hemoglobina aumentada (0,2%), hematocrito aumentado (0,2%), hematocrito disminuido (0,1%), neutrófilos aumentados (0,1%), neutrófilos disminuidos (0,1%), plaquetas aumentadas (0,7%) y plaquetas disminuidas (0,1%). Los porcentajes de los 8119 pacientes que recibieron dosis de gemifloxacino durante un periodo entre 3 días y 14 días (incluso los pacientes que recibieron gemifloxacino durante 5 días) y presentaron resultados anormales de las pruebas de laboratorio con una mayor frecuencia que la observada con 5 días de tratamiento, se citan a continuación: Bioquímica clínica: TGP aumentada (1,7%), TGO aumentada (1,3%), fosfatasa alcalina aumentada (0,4%), uremia aumentada (0,3%), calcio aumentado ( < 0,1%), potasio disminuido (0,1%), sodio disminuido (0,2%), albúmina disminuida (0,3%) y bilirrubina total aumentada (0,4%). Hematología: hemoglobina disminuida (0,2%), hematocrito disminuido (0,3%), glóbulos rojos aumentados (0,1%), glóbulos rojos disminuidos (0,1%), neutrófilos aumentados (0,5%), neutrófilos disminuidos (0,5%), plaquetas aumentadas (1,0%) y plaquetas disminuidas (0,2%). En estudios clínicos de los 8119 pacientes que recibieron 320 mg de gemifloxacino durante un periodo entre 3 días y 14 días (incluso aquellos con tratamiento de 5 días), aproximadamente el 7% de los pacientes tratados con gemifloxacino presentaron valores de TGP elevados inmediatamente antes del ingreso al estudio. De estos pacientes, aproximadamente el 15% mostró un posterior aumento de la TGP en la visita durante el tratamiento y 9% mostró un posterior aumento en la visita del fin del tratamiento. Ninguno de estos pacientes mostró evidencias de ictericia hepatocelular. En cuanto a los comparadores combinados, aproximadamente el 6% presentó valores elevados de TGP inmediatamente antes del ingreso al estudio. De estos pacientes, aproximadamente el 7% mostró una posterior elevación de la TGP en la visita en tratamiento y el 4% mostró una posterior elevación en la visita del fin del tratamiento. En un estudio clínico en que 638 pacientes recibieron una dosis única de 640 mg de gemifloxacino o 250 mg de ciprofloxacino, dos veces por día, durante 3 días, se observó un incremento en la incidencia de los aumentos de la TGP en el grupo del gemifloxacino (3,9%) contra el grupo del comparador (1,0%). En este estudio, dos pacientes experimentaron aumentos de la TGP de 8 a 10 veces el límite superior normal. Estas elevaciones fueron asintomáticas y reversibles. Efectos hepáticos: en los pacientes que reciben 320 mg diarios de gemifloxacino, se producen elevaciones de las enzima hepáticas (aumentos en la TGP y/o la TGO) en proporciones similares a las observadas con agentes antimicrobianos comparadores (ciprofloxacino, levofloxacino, claritro


