MABTHERA®
ROCHE
Denominación genérica: Rituximab.
Forma farmacéutica y formulación: Solución. Cada frasco ámpula de 10 ml contiene: rituximab 100 mg. Vehículo cbp 10 ml. Cada frasco ámpula de 50 ml contiene: rituximab 500 mg. Vehículo cbp 50 ml.
Indicaciones terapéuticas: MABTHERA® está indicado para el tratamiento de pacientes con: linfomas no Hodgkin: linfoma no Hodgkin indolente de linfocitos B, CD20 positivo, en recaída o quimiorresistente o folicular. Linfoma folicular con pacientes en fase III y IV sin tratamiento previo, en combinación con quimioterapia. Como terapia de mantenimiento en pacientes con Linfoma folicular que hayan respondido a la terapia de inducción. Linfoma No Hodgkin difuso de células B grandes, CD20 positivos, en combinación con quimioterapia CHOP (ciclofosfamida, doxorubicina, vincristina y prednisona). Leucemia linfocítica crónica: MABTHERA® está indicado para el tratamiento de leucemia linfocítica crónica (LLC) en primera línea en combinación con quimioterapia. Artritis reumatoide: MABTHERA® en asociación con metotrexato está indicado para el tratamiento de pacientes adultos con artritis reumatoide activa que no hayan respondido adecuadamente al tratamiento con inhibidores del factor de necrosis tumoral (FNT) o no lo toleren.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: mecanismo de acción: rituximab es un anticuerpo monoclonal quimérico de ratón/humano que se une específicamente al antígeno CD20. Este antígeno está localizado en los linfocitos pre-B y B maduros, pero no en las células madre hemopoyéticas, células pro-B, células plasmáticas normales u otros tejidos normales. El antígeno se expresa en > 95% de todos los linfocitos B de los pacientes con linfoma No-Hodgkin (LNH). Posteriormente a la unión a los anticuerpos, el CD20 no es internalizado o proyectado de la membrana celular al medio ambiente. El CD20 no circula en el plasma como antígeno libre y, por lo tanto, no compite por la unión al anticuerpo. Rituximab se une al antígeno CD20 en los linfocitos B e inicia las reacciones inmunológicas que median la lisis de los linfocitos B. Los posibles mecanismos de la lisis celular incluyen citotoxicidad dependiente del complemento (CDC), citotoxicidad celular dependiente del anticuerpo (ADCC) e inducción a la apoptosis. Adicionalmente, los estudios in vitro han demostrado que MABTHERA® (rituximab) sensibiliza a las líneas de linfocitos B del linfoma humano resistentes a fármacos hacia los efectos citotóxicos de algunos agentes quimioterapéuticos. El recuento de células B periféricas descendió hasta valores inferiores a lo normal tras la primera dosis de MABTHERA. En los pacientes con neoplasias hematológicas, la recuperación de la cifra de células B comenzó dentro de los 6 meses siguientes al inicio del tratamiento, normalizándose al cabo de 9-12 meses de concluido éste. En los pacientes con artritis reumatoide, la duración de la depleción de células B varió de unos a otros. La mayoría de ellos continuaron recibiendo tratamiento antes de la recuperación plena de las células B. De los 67 pacientes evaluados para determinar la presencia del anticuerpo humano antimurino (HAMA), ninguno fue positivo. De los 356 pacientes evaluados para anticuerpos humanos antiquiméricos (HACA), el 1,1% (4 pacientes) fueron positivos. Eficacia: linfoma No-Hodgkin folicular o de bajo grado. Monoterapia: tratamiento inicial, con 4 dosis (una por semana). En un estudio fundamental, 166 pacientes con linfoma No-Hodgkin de bajo grado en recaída o quimiorresistente, folicular de linfocitos B, recibieron 375 mg/m2 de MABTHERA®(rituximab) como infusión IV semanalmente en cuatro dosis. La tasa de respuesta global (TRG) en la población de estudio, fue del 48% (IC95% 41%-56%) con una respuesta completa (RC) del 6% y una tasa de respuesta parcial (RP) del 42%. El tiempo promedio hacia la progresión (TPP) para los pacientes que respondieron, fue de 13 meses. En un análisis de subgrupo, la TRG fue más elevada en los pacientes con subtipos histológicos B, C y D de la clasificación "Internacional Working Formulation" (IWF), en comparación a subtipo A de la IWF (58% versus 12%); más elevado en los pacientes cuya lesión fue de < 5cm versus > 7cm en su diámetro más grande (53% versus 38%) y más elevada en los pacientes con recaída quimiosensible en comparación a la recaída quimiorresistente (definido como la duración de respuesta < 3 meses) (50% versus 22%). La TRG en pacientes tratados previamente con trasplante autólogo de médula ósea (TAMO) fue del 78% versus 43% en pacientes sin TAMO. Ni la edad, sexo, grado de linfoma, diagnóstico inicial, presencia o ausencia de enfermedad voluminosa, niveles de DHL normales o elevados, ni la presencia de enfermedad extranodal tienen un efecto estadísticamente significativo (prueba exacta de Fisher) en la respuesta de MABTHERA®. Se observó una correlación estadísticamente significativa entre la tasa de respuesta y el compromiso de la médula ósea. 40% de los pacientes con médula ósea comprometida respondieron comparado con el 59% de los pacientes sin compromiso de la médula ósea (p=0,0186). Este hallazgo no está corroborado por un análisis de regresión logística, en el cual se identificaron los siguientes factores pronósticos: tipo histológico, positividad Bcl-2 basal, resistencia a la última quimioterapia y enfermedad voluminosa. Tratamiento inicial, con 8 dosis (una por semana): en un estudio multicéntrico de un solo brazo, 37 pacientes con linfoma No-Hodgkin (LNH) de bajo grado o folicular de células B reincidentes o quimiorresistentes, recibieron ocho dosis de 375 mg/m2 de MABTHERA como infusión intravenosa. El TRG fue 57% (IC95% 41% - 73%; RC 14%, RP 43%) con un TPP medio para los pacientes que respondieron de 19,4 meses (rango 5,3 a 38,9 meses). Tratamiento inicial de la enfermedad voluminosa, con 4 dosis (una por semana): en datos reunidos a partir de tres estudios, 39 pacientes con LNH de bajo grado o folicular de células B reincidentes o quimiorresistentes, enfermedad voluminosa (lesión única 10 cm en diámetro), recibieron cuatro dosis de 375 mg/m2 de MABTHERA® como infusión intravenosa. La TRG fue de 36% (IC95% 21% - 51%; RC 3%, RP 33%) con un TPP medio para los pacientes que respondieron de 9,6 meses (rango 4,5 a 26,8 meses). Retratamiento, con 4 dosis (una por semana): en un estudio multicéntrico de un solo brazo, 58 pacientes recibieron 375 mg/m2 de MABTHERA® en infusión IV cada semana por cuatro dosis. Todos los pacientes tenían LNH folicular de células B de bajo grado en recaída o refractario y habían alcanzado una respuesta clínica objetiva a un curso previo de MABTHERA®. Tres de los pacientes habían recibido dos cursos de MABTHERA® antes de entrar al estudio y por lo tanto recibieron un tercer curso en este estudio. Dos de los pacientes fueron retratados dos veces en el estudio. Para los 60 retratamientos en estudio, el TRG fue de 38% (IC95% 26% - 51%; 10% RC, 28% RP) con un TPP medio de 17.8 meses (rango 5,4-26,6). Esto se comparó favorablemente con el TPP alcanzado después del primer tratamiento con MABTHERA® (12,4 meses). En combinación con quimioterapia: tratamiento inicial: en un estudio abierto y aleatorio, un total de 322 pacientes con linfoma folicular que no habían recibido tratamiento previo fueron distribuidos aleatoriamente para recibir, bien quimioterapia a base de CVP (750 mg/m2de ciclofosfamida, 1,4 mg/m2 de vincristina hasta un máximo de 2 mg en el día 1 y 40 mg/m2/día de prednisolona en los días 1 a 5) cada 3 semanas por 8 ciclos, o 375 mg/m2de MABTHERA® en combinación con CVP (R-CVP). MABTHERA® se administró en el primer día de cada ciclo de tratamiento. Un total de 321 pacientes (162 R-CVP, 159 CVP) recibieron tratamiento y fueron analizados para determinar la eficacia. La mediana de seguimiento de pacientes fue de 53 meses. R-CVP dio lugar a un beneficio significativo sobre CVP para el indicador primario del estudio, tiempo hasta la falla del tratamiento (27 meses versus 6,6 meses, p < 0,0001, prueba logarítmica). La proporción de pacientes con una respuesta tumoral (RC, RCu, RP) fue significativamente mayor (p < 0.0001, prueba de Chi cuadrada) en el grupo R-CVP (80.9%) que en el grupo CVP (57,2%). El tratamiento con R-CVP prolongó significativamente el tiempo de progresión de la enfermedad o muerte comparado con CVP, 33,6 y 14,7 meses, respectivamente (p < 0,0001, prueba logarítmica). La duración media de respuesta fue de 37,7 meses en el grupo con R-CVP y de 13,5 meses en el grupo de CVP (p < 0,0001, prueba logarítmica). La diferencia entre grupos de tratamiento con respecto a la sobrevida global mostró un beneficio clínico fuerte (p=0,029, prueba logarítmica estratificada por centro): la tasa de sobrevida a los 53 meses fue de 80.9% para pacientes en el grupo de R-CVP comparado con 71,1% para pacientes en el grupo CVP. Los resultados de otros tres estudios aleatorios que utilizaron MABTHERA® en combinación con un régimen de quimioterapia distinto a CVP (CHOP, MCP, CHVP/Interferón-a) también demostraron mejoría significativa en las tasas de respuesta, parámetros dependientes del tiempo así como en la sobrevida global. Los resultados clave provenientes de los cuatro estudios se resumen en la siguiente tabla.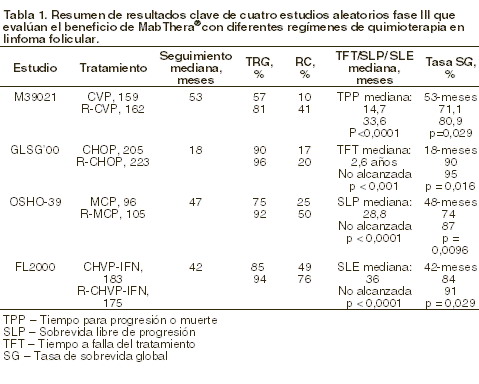
Terapia de mantenimiento: en un estudio fase III, prospectivo, abierto, internacional y multicéntrico, 465 pacientes con LNH folicular recurrente o refractario fueron distribuidos aleatoriamente en una primera fase a recibir tratamiento de inducción con CHOP (n = 231) o MABTHERA® más CHOP (R-CHOP, n = 234). Los dos grupos estaban bien equilibrados en cuanto a características basales y estado de la enfermedad. En una segunda fase, se asignó aleatoriamente a un total de 334 pacientes con remisión completa o parcial tras la terapia de inducción para recibir tratamiento de mantenimiento con MABTHERA® (n = 167) o permanecer solamente en observación (n = 167). El tratamiento de mantenimiento con MABTHERA® consistió en una infusión a dosis de 375 mg/m2 cada 3 meses hasta la progresión de la enfermedad o durante un período máximo de 2 años. El análisis final de eficacia incluyó a todos los pacientes asignados aleatoriamente a ambas partes del estudio. Después de una mediana de observación de 31 meses de los pacientes asignados aleatoriamente en la fase de inducción, los resultados de los pacientes con LNH folicular recurrente o refractario del grupo que recibió R-CHOP mejoraron significativamente en comparación con el grupo de quimioterapia (ver tabla 2)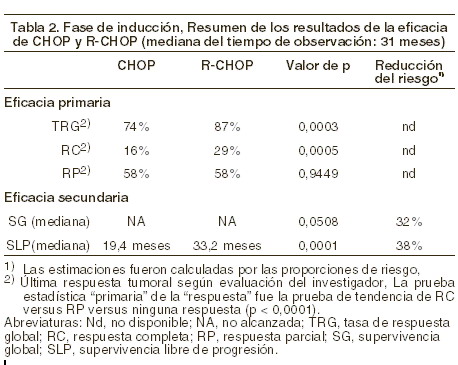
Para los pacientes asignados aleatoriamente en la fase de mantenimiento del estudio, la mediana del tiempo de observación fue de 28 meses desde la asignación aleatoria. La terapia de mantenimiento con MABTHERA® se tradujo en una mejoría estadísticamente significativa y clínicamente importante de la variable de eficacia primaria, SLP (tiempo entre la asignación aleatoria para el mantenimiento y la recaída, la progresión de la enfermedad y la muerte), en comparación con el grupo de sólo observación (p < 0,0001, prueba de rangos logarítmicos). La mediana de la SLP fue de 42,2 meses en el grupo de mantenimiento con MABTHERA® versus 14,3 meses en el grupo de observación. Aplicando un análisis de regresión de Cox, el riesgo de enfermedad progresiva o muerte disminuyó en un 61% en el grupo de terapia de mantenimiento con MABTHERA® en comparación con el grupo de observación (IC del 95%; 45-72%). La tasa estimada de sobrevida libre de progresión a los 12 meses según el método de Kaplan-Meier fue del 78% en el grupo de mantenimiento con MABTHERA® versus 57% en el grupo de observación. Un análisis de sobrevida global confirmó los beneficios significativos del mantenimiento con MABTHERA® sobre la observación (p = 0,0039, prueba de rangos logarítmicos). La terapia de mantenimiento con MABTHERA® redujo el riesgo de muerte en un 56% (IC del 95%, 22-75%). La mediana de la duración hasta un nuevo tratamiento para el antilinfoma fue significativamente más larga en el grupo de terapia de mantenimiento con MABTHERA® que en el de observación (38,8 versus 20,1 meses; p < 0,0001, prueba de rangos logarítmicos). El riesgo de empezar un nuevo tratamiento disminuyó en un 50% (IC del 95%, 30-64%). En los pacientes con una RC/RCnc (respuesta completa no confirmada) como mejor respuesta durante el tratamiento de inducción, la terapia de mantenimiento con MABTHERA® prolongó significativamente la mediana de la sobrevida libre de enfermedad (SLE) en comparación con el grupo de observación (53,7 versus 16,5 meses; p = 0,0003, prueba de rangos logarítmicos) (ver tabla 3). El riesgo de recaída de los pacientes con respuesta completa fue del 67% (IC del 95%, 39-82%).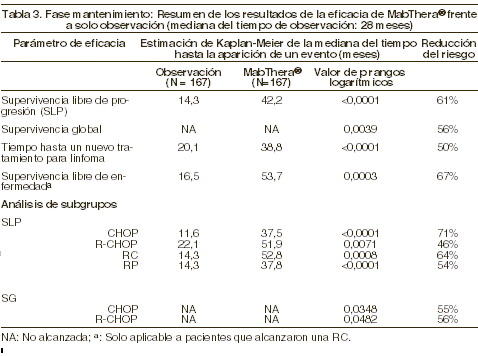
El beneficio del tratamiento con MABTHERA® se confirmó en todos los subgrupos analizados, independientemente del régimen de inducción (CHOP o R-CHOP) o la calidad de la respuesta al tratamiento de inducción (RC o RP) (ver tabla 3). La terapia de mantenimiento con MABTHERA® prolongó significativamente la SLP en los pacientes que respondieron al tratamiento de inducción con CHOP (mediana de SLP: 37,5 versus 11,6, p < 0,0001), así como en los que respondieron a la inducción con R-CHOP (mediana de SLP: 51,9 versus 22,1 meses, p = 0,0071). El tratamiento de mantenimiento con MABTHERA® también aportó beneficios clínicamente significativos en cuanto a sobrevida global tanto para los pacientes que respondieron a CHOP como para los que lo hicieron a R-CHOP en la fase de inducción del estudio. La terapia de mantenimiento con MABTHERA® se tradujo en beneficios consistentes para todos los subgrupos de estudio [género (masculino, femenino)], edad (≤60 años, > 60 años), estadio (III, IV), estado de desempeño según la OMS (0 versus > 0), síntomas B (ausentes, presentes), afectación de la médula ósea (no versus si), IPI (0-2 versus 3-5), FLIPI (0-1 versus 3-5), número de sitios extranodales (0-1 versus > 1), número de sitios nodales ( < 5 versus ≥ 5), número de regímenes terapéuticos anteriores (1 versus 2), mejor respuesta a tratamiento previo (RC/RP versus sin cambios/EP: enfermedad progresiva), hemoglobina ( < 12 g/dl frente a ≥ 12 g/dl), b2-microglobulina ( < 3 mg/l versus ≥ 3 mg/dl), DHL (elevada, no elevada) excepto para el pequeño subgrupo de pacientes con enfermedad voluminosa. Linfoma no Hodgkin difuso de células B grandes (LDCBG): en un estudio aleatorio, abierto, un total de 399 pacientes ancianos no tratados previamente (edad de 60 a 80 años), con linfoma difuso de células B grandes recibieron quimioterapia CHOP estándar (ciclofosfamida 750 mg/m2, doxorubicina 50 mg/m2, vincristina 1,4 mg/m2hasta un máximo de 2 mg en el día 1 y prednisolona 40 mg/m2/día en los días 1-5), cada 3 semanas durante ocho ciclos, o MABTHERA® 375 mg/m2 más CHOP (R-CHOP). MABTHERA® fue administrado en el primer día del ciclo de tratamiento. El análisis de eficacia incluyó todos los pacientes asignados aleatoriamente (197 CHOP, 202 R-CHOP) y tuvo un seguimiento promedio de 31 meses de duración. Los dos grupos tratados fueron bien balanceados en las características basales y el estatus de la enfermedad. El análisis final confirmó que R-CHOP incrementó significativamente la duración de la sobrevida libre de evento (parámetro principal de la eficacia en donde los eventos fueron muerte, recaída, progresión del linfoma, o aplicación de un tratamiento anti-linfoma) (p = 0,0001). El análisis de Kaplan-Meier estima que la duración media de la sobrevida libre de evento fue de 35 meses en el grupo R-CHOP comparado con 13 meses en el grupo CHOP, representando una reducción del riesgo de 41%. A los 24 meses, la sobrevida estimada fue de 68,2% en el grupo de R-CHOP y del 57.4% en el grupo CHOP. Un análisis subsecuente de la duración de la sobrevida total llevada a cabo con un seguimiento medio de 60 meses, confirmó el beneficio de R-CHOP sobre el tratamiento CHOP (p = 0,0071) con una reducción del riesgo de 32%. El análisis de los parámetros secundarios (tasas de respuesta, sobrevida libre de progresión, sobrevida libre de enfermedad, duración de la respuesta) confirmó el efecto del tratamiento de R-CHOP comparado con CHOP. La tasa de respuesta completa después del 8vo ciclo fue 76,2% en el grupo R-CHOP y 62,4% en el grupo CHOP (p=0,0028). El riesgo de progresión de la enfermedad se redujo cerca de un 46% y el riesgo de recaída cerca de un 51%. En todos los subgrupos de pacientes (género, edad, IPI ajustado por edad, estadio Ann Arbor, ECOG, Beta 2 microglobulina, DHL, albúmina, síntomas B, enfermedad nodular, sitios extranodales, afección de la médula espinal), el riesgo relativo para sobrevida libre de evento y la sobrevida total (R-CHOP comparado con CHOP) fueron menores del 0,83 y 0,95 respectivamente. R-CHOP se asoció a mejores tasas de respuesta en pacientes con IPI ajustado a la edad tanto alto como bajo. Leucemia linfocítica crónica: en un estudio aleatorizado abierto, un total de 817 pacientes sin tratamiento previo con LLC fueron asignados de manera aleatoria para recibir quimioterapia FC (fludarabina 25 mg/m2, ciclofosfamida 250 mg/m2, los días 1-3) cada cuatro semanas durante 6 ciclos o MABTHERA® en combinación con FC (R-FC). Se administró MABTHERA® a una dosis de 375 mg/m2 durante el primer ciclo un día antes de la quimioterapia y a una dosis de 500 mg/m2 el día 1 de cada ciclo subsecuente de tratamiento. Se analizó un total de 810 pacientes (403 R-FC, 407 FC) para evaluar eficacia. La variable primaria de sobrevida libre de progresión tuvo una media de 40 meses para el grupo R-FC y 32 meses en el grupo FC (p < 0,0001, rango logarítmico). El análisis global de sobrevida mostró una mejora en la sobrevida a favor del grupo R-FC (p = 0,0427), sin embargo es necesario un seguimiento más largo para confirmar esta observación. El beneficio en términos de SLP fue consistentemente observado en la mayoría de subgrupos de pacientes analizados de acuerdo con el riesgo de enfermedad en la medición basal.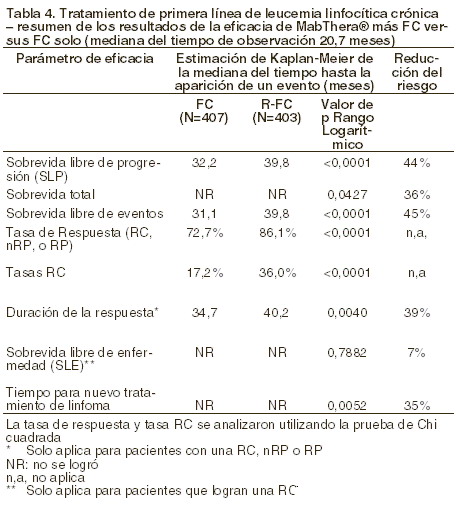
Los resultados de otros estudios de soporte utilizando MABTHERA® en combinación con otros regímenes de quimioterapia (incluyendo CHOP, FCM, PC, PCM) para el tratamiento de pacientes con LLC también han demostrado altas tasas de respuesta con tasas totales de SLP prometedoras sin agregar toxicidad importante al tratamiento. Artritis reumatoide: la eficacia y la seguridad de MABTHERA para aliviar los signos y síntomas de artritis reumatoide quedaron demostradas en tres estudios multicéntricos, aleatorios, doble ciego, controlados. El estudio 1 fue un ensayo clínico comparativo doble ciego, en el que participaron 517 pacientes que no habían respondido adecuadamente a uno o más tratamientos con inhibidores del FNT o no lo toleraban. Los pacientes elegibles presentaban artritis reumatoide activa grave, diagnosticada de acuerdo a los criterios del colegio americano de reumatología (ACR, por sus siglas en inglés: American College of Rheumatology). La variable principal era el porcentaje de pacientes con una respuesta ACR20 en la semana 24. Los pacientes recibieron dos infusiones IV de 1000 mg de MABTHERA® cada una de ellas tras una infusión IV de 100 mg de metilprednisolona y separadas por un intervalo de 15 días. Todos los pacientes recibieron concomitantemente metotrexato oral (10-25 mg/semana), así como 60 mg de prednisolona oral los días 2-7 y 30 mg los días 8-14 tras la primera infusión. Los pacientes recibieron seguimiento después de la semana 24 para evaluación a largo plazo, incluyendo evaluación radiográfica a la semana 56. Durante este período, los pacientes pudieron haber recibido dosis adicionales de rituximab bajo un esquema de extensión de protocolo abierto. El estudio 2 fue un ensayo clínico aleatorizado, doble ciego, controlado con doble placebo y multifactorial 3x3, en el que se compararon dos dosis diferentes de rituximab administrado con o sin uno de dos regímenes de corticosteroides en infusión, en asociación con metotrexato semanalmente, en pacientes con artritis reumatoide activa que no habían respondido al tratamiento con al menos otros 5 FARME (fármacos antirreumáticos modificadores de la enfermedad). El estudio 3 fue un ensayo clínico doble ciego, controlado con doble placebo, en el que se evaluó rituximab en monoterapia y en asociación con ciclofosfamida o metotrexato en pacientes con artritis reumatoide activa que no habían respondido antes a uno o más FARME. El grupo de comparación en los tres estudios recibió metotrexato semanalmente (10-25 mg/semana). Parámetros de la actividad de la enfermedad: en los tres estudios, la administración de 2 veces 1.000 mg de rituximab incrementó la proporción de pacientes con una mejoría del índice ACR de un 20% como mínimo en comparación con los pacientes tratados con metotrexato solo (ver tabla 5). El efecto terapéutico fue similar con independencia de variables como factor reumatoide, edad, sexo, superficie corporal, raza, número de tratamientos anteriores o estado clínico de los pacientes. También se observó una mejoría clínica y estadísticamente significativa de todos los componentes de la respuesta ACR (número de articulaciones dolorosas e inflamadas, evaluación global del paciente y del médico), índice HAQ [medida de la incapacidad funcional], evaluación del dolor y proteína C reactiva (mg/dl).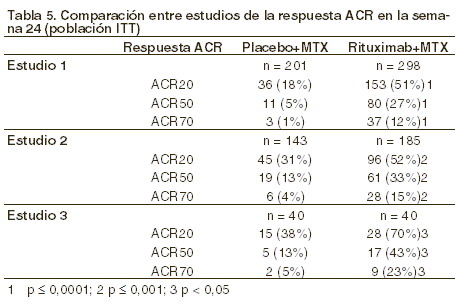
En el estudio 3, la respuesta ACR20 en los pacientes tratados con rituximab solo fue del 65%, frente al 38% en los que recibieron metotrexato solo (p = 0,025). En los pacientes tratados con rituximab fue significativamente mayor el descenso del índice de actividad de la enfermedad (DAS28) que en los que recibieron metotrexato solo. Un número significativamente mayor de pacientes tratados con rituximab alcanzó una respuesta EULAR buena o moderada en comparación con los pacientes tratados con metotrexato solo (ver tabla 6).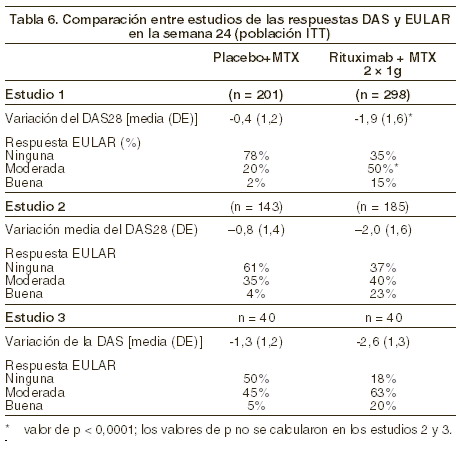
Respuesta radiológica: el estudio 1 fue realizado en pacientes con respuesta inadecuada o intolerancia a una o más terapias con inhibidores del FNT, el daño articular estructural fue evaluado radiológicamente y expresado como cambios en la escala total modificada de Sharp y sus componentes, la escala de erosión y la escala de estrechamiento del espacio articular. En la semana 56, los pacientes del grupo rituximab/MTX demostraron una disminución significativa de la progresión radiológica en comparación con los pacientes del grupo de metotrexato solo. Una mayor proporción de pacientes con rituximab no presentaron progresión erosiva después de la semana 56 (ver tabla 7).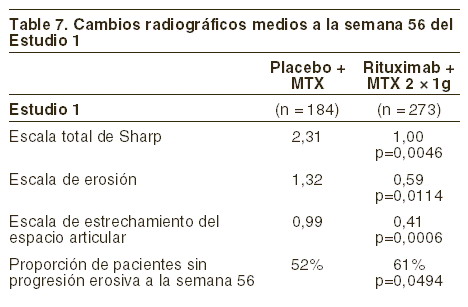
Parámetros de la calidad de vida: los pacientes tratados con rituximab notificaron una mejoría de todos los parámetros autoevaluados (cuestionarios HAQ-DI, FACIT-F y SF-36, ver tablas 8 y 9). Se alcanzaron reducciones significativas en las puntuaciones de incapacidad (HAQ-DI) y fatiga (FACIT-F), así como una mejoría de los aspectos sobre salud física y mental del cuestionario SF-36 en los pacientes tratados con rituximab en comparación con los que recibieron metotrexato solo.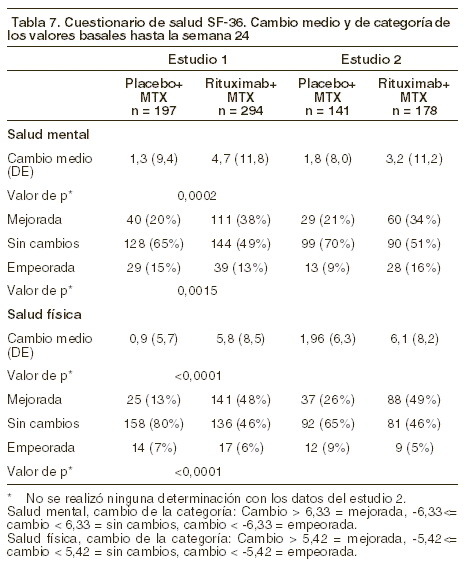
En la semana 24 y en los tres estudios, la proporción de pacientes tratados con rituximab con una mejoría clínicamente relevante en el HAQ-DI (definida como descenso de la puntuación total individual de > 0,25) fue mayor que entre los pacientes tratados con metotrexato solo. Evaluaciones de laboratorio: en los estudios clínicos, el número de pacientes con artritis reumatoide que presentaban anticuerpos antiquiméricos humanos (HACA, por sus siglas en inglés: Human anti-chimeric antibodies) fue aproximadamente del 10%. La detección de HACA no fue asociada con deterioro clínico ni con incremento en el riesgo de reacciones a infusiones subsecuentes en la mayoría de los pacientes. La presencia de HACA podría ser asociada con el empeoramiento de las reacciones alérgicas asociadas a la infusión después de la segunda infusión o dosis subsecuentes; rara vez se ha observado la falla en la depleción de células B posterior a la aplicación de dosis adicionales de tratamiento. Entre los pacientes que dieron positivo al análisis de factor reumatoide (FR) se observaron descensos pronunciados de las concentraciones de FR tras el tratamiento con rituximab en los tres estudios (intervalo: 45-64%, ver figura 1).
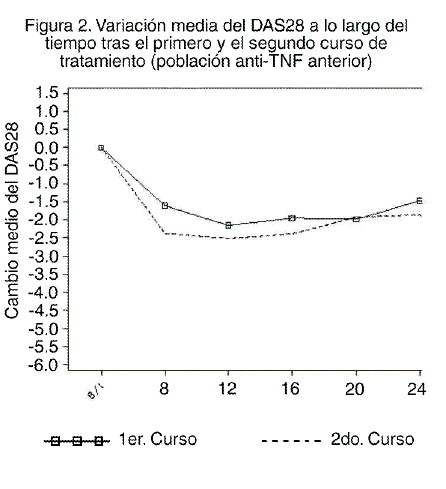
Las concentraciones plasmáticas de inmunoglobulinas totales, los recuentos totales de linfocitos y las cifras de leucocitos se mantuvieron en general dentro de los límites normales tras la administración de MABTHERA®, con excepción de una caída temporal en el número de leucocitos en las primeras cuatro semanas tras el tratamiento. Los títulos de anticuerpos específicos IgG contra antígenos de paperas, rubéola, varicela, toxoide tetánico, gripe y Streptococcus pneumococci permanecieron estables durante 24 semanas tras la exposición a MABTHERA® en los pacientes con artritis reumatoide. Los efectos del rituximab en diversos biomarcadores se evaluaron en los pacientes participantes en el estudio 3. En este subestudio se evaluó el impacto de un curso único de rituximab en los niveles de marcadores bioquímicos, a saber: marcadores de inflamación [interleucina 6, proteína C reactiva, proteína SAA (suero amiloide A), proteína S100 isotipos A8 y A9], autoanticuerpos (FR y anticuerpos anti-péptidos cíclicos citrulinados) producción y recambio óseo [osteocalcina y péptido amino-terminal del procolágeno 1 (P1NP)]. El tratamiento con rituximab, en monoterapia o en asociación con metotrexato o ciclofosfamida, redujo los niveles de marcadores inflamatorios significativamente en comparación con el metotrexato solo durante las primeras 24 semanas de seguimiento. Los niveles de marcadores de recambio óseo, osteocalcina y P1NP aumentaron significativamente con rituximab en comparación con metotrexato solo. Cursos múltiples de tratamiento: terminado el período de estudio comparativo doble ciego de 24 semanas, los pacientes podían participar en un estudio abierto de seguimiento a largo plazo. Los pacientes recibieron nuevos cursos de MABTHERA®según las necesidades de acuerdo con la evaluación por el médico de la actividad de la enfermedad y sin tener en cuenta la cifra de linfocitos B periféricos. El lapso entre dos cursos fue variable, pero la mayoría de los pacientes fue tratado de nuevo a los 6-12 meses de finalizado el curso anterior. La necesidad de re-tratamiento fue incluso menos frecuente en algunos pacientes. Cuando se continuó el tratamiento, la respuesta fue al menos de igual magnitud que tras el curso de tratamiento inicial, como mostraba la variación del DAS28 (ver figura 2).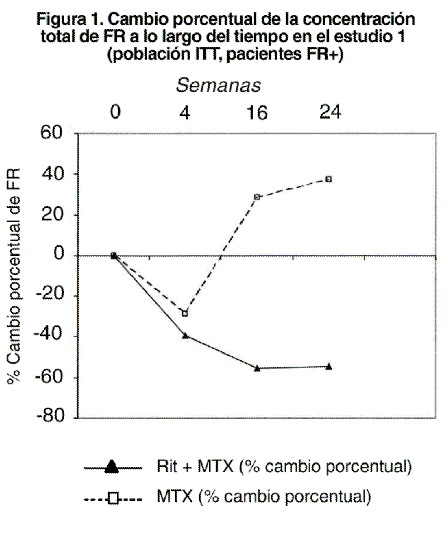
Propiedades farmacocinéticas: eliminación y distribución: linfomas no-Hodgkin: basado en un análisis farmacocinético de población en 298 pacientes LNH que recibieron infusiones simples o múltiples de rituximab como agente solo o en combinación con terapia CHOP, los típicos cálculos de depuración no específica (CL1), depuración específica (CL2) de la población probablemente contribuidas por la carga de células B o tumoral y el volumen de distribución central compartimental (V1) fueron de 0,14 l/día, 0,59 l/día y 2,7 l, respectivamente. La vida media de eliminación promedio estimada del rituximab fue de 22 días (rango, 6,1 a 52 días). La cuenta basal de células CD-19 positivas y el tamaño de lesiones tumorales medibles contribuyeron a algo de variabilidad en el CL2 de rituximab en datos de 161 pacientes a los que se les administró 375 mg/m2 como infusión IV por 4 dosis semanales. Los pacientes con mayores cuentas de células CD-19 positivas o lesiones tumorales tuvieron una mayor CL2. Sin embargo, se mantuvo un gran componente de variabilidad interindividual para el CL2 después de la corrección para la cuenta de células CD-19 positivas y tamaño de lesión tumoral. El V1 varió por el área de superficie corporal (ASC) y la terapia con CHOP. Esta variabilidad en el V1 (27,1% y 19,0%) contribuida por el rango del ASC (1,53 a 2,32 m2) y la terapia concurrente con CHOP, respectivamente, fue relativamente pequeña. La edad, género, raza y estatus de desempeño de la OMS no tuvieron efecto sobre la farmacocinética de rituximab. Este análisis sugiere que el ajuste de dosis de rituximab con cualquier co-variable probada no se espera que provoque una reducción significativa en su variabilidad farmacocinética. Rituximab a una dosis de 375 mg/m2 se administró como infusión IV a intervalos semanales para 4 dosis a 203 pacientes con LNH sin tratamiento previo con rituximab. La Cmáx promedio después de la cuarta infusión fue de 486 mg/ml (rango, 77,5 a 996,6 mg/ml). Los niveles séricos máximos y mínimos de rituximab se correlacionaron de manera inversa con valores basales para el número de células B CD19 positivas circulantes y las mediciones de carga de enfermedad. Los niveles séricos en el estado estacionario fueron mayores para los pacientes con respuesta en comparación con los de no respuesta. Los niveles séricos fueron mayores en pacientes con formulación internacional de trabajo (FIT) subtipos B, C y D en comparación con aquellos con subtipo A. Rituximab fue detectable en el suero de pacientes de 3 a 6 meses después de completar el último tratamiento. Rituximab a una dosis de 375 mg/m2 se administró como infusión IV a intervalos semanales por 8 dosis a 37 pacientes con LNH. La Cmáxpromedio se incrementó con cada infusión sucesiva, expandiéndose de un promedio de 243 mg/ml (rango, 16-582 mg/ml) después de la primera infusión a 550 mg/ml (rango, 171-1.177 mg/ml) después de la octava infusión. El perfil farmacocinético de rituximab al administrarse como 6 infusiones de 375 mg/m2 en combinación con 6 ciclos de quimioterapia con CHOP fue similar a la observada con rituximab solo. Artritis reumatoide: tras la administración de dos infusiones IV de rituximab de 1.000 mg cada una, separadas por dos semanas, la vida media terminal fue de 20,8 días (intervalo: 8,58-35,9 días); la depuración sistémica media, de 0,23 l/día (intervalo: 0,091-0,67 l/día) y el volumen medio del estado estacionario fue de de 4,6 l (intervalo: 1,7-7,51 l). El análisis farmacocinético de poblaciones de los mismos datos arrojó unos valores medios similares para la depuración sistémica y la vida media: 0,26 l/día y 20,4 días, respectivamente. El análisis farmacocinético de poblaciones reveló que el área de la superficie corporal (ASC) y el sexo fueron las covariables más significativas para explicar la variabilidad interindividual de los parámetros farmacocinéticos. Tras el ajuste por el ASC, el volumen de distribución era mayor y la depuración más rápida en los sujetos masculinos que en los femeninos. Las diferencias farmacocinéticas asociadas al sexo no se consideraron clínicamente importantes y, por tanto, no es necesario ajustar la dosis. Tras la administración intravenosa de dosis de 500 y 1.000 mg de rituximab en dos ocasiones, separadas por dos semanas, los valores medios de Cmáx eran de 183 g/ml (intervalo: 81,8-279 mg/ml) y 370 mg/ml (212-637 mg/ml), y los de la vida media, de 17,9 días (intervalo: 12,3-31,3 días) y 19.7 días (intervalo: 12,3-34,6 días), respectivamente. No existen datos farmacocinéticos sobre los pacientes tratados con cursos múltiples de tratamiento. Después de recibir la misma pauta de administración (2 veces 1.000 mg IV, separadas por dos semanas), los parámetros farmacocinéticos en los pacientes con una respuesta inadecuada al tratamiento con inhibidores del FNT fueron similares, con una concentración sérica media máxima de 369 mg/ml y una vida media terminal de 19,2 días. Farmacocinética en poblaciones especiales: no existen datos farmacocinéticos de pacientes con insuficiencia hepática o renal.
Contraindicaciones: MABTHERA® está contraindicado en los pacientes con hipersensibilidad conocida a rituximab, o a cualquiera de los componentes del producto o a las proteínas murinas.
Precauciones generales: Pacientes con linfoma no-Hodgkin y leucemia linfocítica crónica: las infusiones de MABTHERA® deben administrarse en un medio en donde se encuentren disponibles las instalaciones para una resucitación completa y bajo la estrecha supervisión de un oncólogo/hematólogo experimentado. Reacciones relacionadas con la infusión: MABTHERA® está asociado con reacciones vinculadas a la infusión, las cuales pueden estar relacionadas a la liberación de citocinas y/u otros mediadores químicos. Las reacciones severas relacionadas a la infusión pueden ser clínicamente indistinguibles de las reacciones de hipersensibilidad o del síndrome de liberación de citocinas. Durante el uso del medicamento en el período de postcomercialización, se han reportado reacciones severas relacionadas a la infusión, con resultados fatales. Las reacciones severas relacionadas a la infusión generalmente manifestadas dentro de 30 minutos a 2 horas después de iniciar la primera infusión de MABTHERA®, se caracterizaron por eventos pulmonares, e incluyeron, en algunos casos lisis tumoral rápida y características del síndrome de lisis tumoral, además de fiebre, escalofríos, rigidez, hipotensión, urticaria, angioedema y otros síntomas (ver Reacciones secundarias y adversas). Los pacientes con una elevada carga tumoral o con un número elevado ( > 25 x 109/l) de células malignas circulantes, tales como pacientes con leucemia linfocítica crónica (LLC) y linfoma del manto celular pueden estar en riesgo elevado de desarrollar reacciones severas relacionadas con la infusión. Los síntomas de reacciones a la infusión generalmente son reversibles con la interrupción de la infusión. Se recomienda el tratamiento de síntomas relacionados con la infusión con difenhidramina y acetaminofen. Puede estar indicado un tratamiento adicional con broncodilatadores o solución salina IV. En la mayoría de los casos, la infusión se puede reiniciar a un 50% de velocidad (por ejemplo, de 100 mg/h a 50 mg/h) cuando se hayan resuelto todos los síntomas. La mayoría de los pacientes que han experimentado reacciones relacionadas con la infusión que no ponen en riesgo su vida han podido completar el curso completo de la terapia con MABTHERA®. Un tratamiento posterior de pacientes después de la completa resolución de signos y síntomas ha provocado en raras ocasiones reacciones severas repetidas relacionadas con la infusión. Se han reportado reacciones anafilácticas y otras reacciones de hipersensibilidad después de la administración intravenosa de proteínas a pacientes. Se debe tener epinefrina, antihistamínicos y glucocorticoides disponibles para uso inmediato en el caso de alguna reacción de hipersensibilidad a MABTHERA®. Pacientes con un alto número ( > 25 x 109/l) de células malignas circulantes o alta carga tumoral tales como los pacientes con LLC y linfoma de las células del manto, quienes pueden estar a un mayor riesgo de reacciones especialmente severas relacionadas con la infusión, deben tratarse solamente con extrema precaución. Estos pacientes deben monitorearse de manera muy cercana a lo largo de la primera infusión. Se debe tener en cuenta el uso de una velocidad de infusión reducida para la primera infusión en estos pacientes o dividir la dosis en dos días durante el primer ciclo. Eventos pulmonares: los eventos pulmonares han incluido hipoxia, infiltrados pulmonares e insuficiencia respiratoria aguda. Algunos de estos eventos fueron precedidos por broncoespasmo severo y disnea. En algunos casos, los síntomas empeoraron con el tiempo mientras que en otros la mejoría inicial fue seguida por el deterioro clínico. Por lo tanto, los pacientes que experimenten eventos pulmonares u otros síntomas severos relacionados con la infusión, deben ser monitoreados estrechamente hasta la completa resolución de sus síntomas. Los pacientes con antecedentes de insuficiencia pulmonar o aquellos con infiltración tumoral pulmonar, pueden estar en un mayor riesgo de obtener un resultado pobre y deben ser tratados con mucha precaución. Insuficiencia respiratoria aguda puede acompañarse por eventos tales como infiltración pulmonar intersticial o edema, visible en rayos X del pecho. El síndrome se manifiesta generalmente a no más de una o dos horas de iniciar la primera infusión. A los pacientes que experimenten eventos pulmonares severos se les debe interrumpir inmediatamente su infusión (ver Dosis y vía de administración) y deben recibir tratamiento sintomático agresivo. Lisis tumoral aguda: MABTHERA® media la lisis rápida de las células CD20 positivas benignas y malignas. Se ha reportado que los signos y síntomas (por ejemplo hiperuricemia, hipercalemia, hipocalcemia, hiperfosfatemia, insuficiencia renal aguda, DHL elevada) consistentes con el síndrome de lisis tumoral (SLT), se presentan después de la primera infusión de MABTHERA®, en los pacientes con números elevados de linfocitos malignos circulantes. La profilaxis para SLT debe considerarse para los pacientes en riesgo de desarrollar lisis tumoral rápida (por ejemplo, pacientes con una alta carga tumoral o con un alto número ( > 25 x 109/l) de células malignas circulantes tales como pacientes con LLC y linfoma de las células del manto). Estos pacientes deben ser seguidos estrechamente y se les debe realizar un adecuado monitoreo de laboratorio. Debe proporcionarse la adecuada terapia médica para los pacientes quienes desarrollan signos y síntomas consistentes con una rápida lisis tumoral. Después del tratamiento para la completa resolución de los signos y síntomas, se debe administrar la subsiguiente terapia con MABTHERA®, en conjunción con una terapia profiláctica para SLT en un número limitado de casos. Cardiovascular: debido a que se puede presentar hipotensión durante la infusión de MABTHERA®, se debe tomar en consideración la suspensión de los medicamentos antihipertensivos 12 horas antes de y durante toda la infusión de MABTHERA®. Angina de pecho o arritmias cardíacas, tales como aleteo auricular y fibrilación, se han presentado en los pacientes tratados con MABTHERA®. Por lo tanto, los pacientes con antecedentes de enfermedad cardíaca deben monitor
earse estrechamente durante las infusiones. Monitoreo de cuenta sanguínea: aún cuando MABTHERA® no es mielosupresor como monoterapia, se debe tener precaución en los pacientes con cuenta de neutrófilos de < 1,5 a 109/l y/o cuenta de plaquetas de < 75 x 109/l, porque la experiencia clínica con dichos pacient