RECORMON®
ROCHE
Denominación genérica: Eritropoyetina beta.
Forma farmacéutica y formulación: Solución inyectable. Jeringas precargadas: cada jeringa precargada con 5.000 UI contiene: eritropoyetina beta 5.000 UI. Vehículo cbp 0,3 ml. Multidosis: cada frasco ámpula con 50.000 UI contiene: eritropoyetina beta humana recombinante 50.000 UI. Ampolleta con diluyente: alcohol bencílico 40 mg, cloruro de benzalconio 0,20 mg. Agua inyectable cbp 10 ml.
Indicaciones terapéuticas: Tratamiento de la anemia asociada a insuficiencia renal crónica (anemia renal) en pacientes sometidos a diálisis peritoneal. Tratamiento de la anemia renal sintomática en pacientes todavía no sometidos a diálisis peritoneal. Prevención de la anemia en prematuros con un peso de 750-1.500 g al nacer y una edad gestacional inferior a 34 semanas. Prevención y tratamiento de la anemia en pacientes adultos con todos los tumores no mieloides, sometidos a quimioterapia. Aumentar el rendimiento de la sangre autóloga en los programas de autotransfusión. El uso en esta indicación debe sopesarse frente al aumento del riesgo de accidentes tromboembólicos. A los pacientes con anemia moderada (Hb: 10-13 g/dl [6,2-8,1 mmol/l] o hematocrito: 30-39%, sin deficiencia de hierro), sólo se les tratará si no se dispone de sistemas para la conservación de sangre o si no bastan ante una intervención de cirugía mayor programada que requiera un alto volumen de sangre (4 o más unidades de sangre para las mujeres y 5 o más para los hombres).
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: según investigaciones farmacocinéticas en voluntarios sanos y pacientes urémicos, la vida media de la eritropoyetina beta administrada por vía IV oscila entre 4 y 12 horas, y el volumen de distribución es entre una y dos veces superior al volumen plasmático. Resultados similares se han alcanzado en los ensayos con ratas urémicas y normales. Tras la administración SC de eritropoyetina beta a pacientes urémicos, su absorción prolongada se traduce en una concentración sérica en equilibrio cuyo valor máximo se alcanza al cabo de 12 a 28 horas. Con una media de 13 a 28 horas, la vida media terminal es mayor que tras la administración intravenosa. Biodisponibilidad: la biodisponibilidad de la eritropoyetina beta por vía SC varía entre el 23 y el 42 % de la alcanzada por vía intravenosa. Propiedades farmacodinámicas: propiedades y efectos: en su composición de aminoácidos y carbohidratos, la eritropoyetina beta es idéntica a la eritropoyetina aislada de la orina de pacientes anémicos. La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de los progenitores eritroides. Actúa como factor estimulante de la mitosis y hormona de diferenciación. La eficacia biológica de la eritropoyetina beta tras su administración por vía intravenosa (IV) y subcutánea (SC) se ha demostrado en diversos modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Después de recibir eritropoyetina beta, aumentaban los recuentos de eritrocitos y reticulocitos, los valores de hemoglobina y la incorporación globular de 59Fe. In vitro (cultivos de células esplénicas de ratón), se ha observado un aumento de la incorporación de 3H-timidina a los núcleos eritroides de células de bazo tras la incubación con eritropoyetina beta. Por otro lado, en cultivos de células de médula ósea humana se ha puesto de manifiesto que la eritropoyetina beta estimula específicamente la eritropoyesis y que no afecta a la leucopoyesis. No se han detectado efectos citotóxicos de la eritropoyetina beta sobre la médula ósea o la piel del ser humano. Tras la administración de una dosis única de eritropoyetina beta, no se ha apreciado ningún cambio del comportamiento y la actividad locomotora del ratón ni de la función respiratoria del perro. La eritropoyetina es un factor de crecimiento que estimula principalmente la producción de células rojas. Los receptores de eritropoyetina pueden ser expresados en la superficie de una variedad de células tumorales. Existe poca información para establecer si el uso de los productos de eritropoyetina tienen un efecto nocivo en el tiempo de la progresión del tumor o en la sobrevida libre de progresión. Dos estudios exploraron el efecto de la eritropoyetina en la sobrevida y/o la progresión del tumor con valores mayores de hemoglobina. En un estudio controlado contra placebo y aleatorizado usando eritropoyetina alfa en 939 pacientes de cáncer de mama metastásico, el fármaco de estudio fue administrado para procurar mantener niveles de la hemoglobina entre 12 y 14 g/dl. A cuatro meses, los casos de muerte atribuidos a la progresión de la enfermedad eran mayores (6% contra 3%) en mujeres que recibieron eritropoyetina alfa. La mortalidad total fue significativamente más alta en el grupo que recibió eritropoyetina alfa. En otro estudio placebo-controlado usando eritropoyetina beta en 351 pacientes con cáncer de cabeza y del cuello, el fármaco de estudio fue administrado para mantener los niveles de hemoglobina de 14 g/dl en mujeres y de 15 g/dl en hombres. La sobrevida libre de progresión fue significativamente más corta en los pacientes que recibían eritropoyetina beta. Los resultados de este estudio fueron afectados por variaciones (sesgo por desequilibrios) entre los grupos del tratamiento, especialmente respecto de la localización del tumor, el hábito tabáquico y a la heterogeneidad de la población del estudio. Además, muchos otros estudios han demostrado una tendencia a la sobrevivencia mejorada que sugería que la eritropoyetina no tiene ningún efecto negativo en la progresión del tumor. En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO. Datos no clínicos de seguridad: datos no clínicos de seguridad revelan ningún peligro especial para los humanos, basados en estudios convencionales de seguridad farmacológica, toxicidad a dosis repetidas, genotoxicidad y toxicidad en la reproducción. Un estudio de carcinogenicidad con eritropoyetina homologa en ratones no reveló signos proliferativos de potencial tumorigénico.
Contraindicaciones: Hipersensibilidad a la sustancia activa o alguno de sus excipientes. Hipertensión mal controlada. Para la indicación "aumentar el rendimiento de la sangre autóloga", RECORMON® no debe utilizarse en pacientes que hayan sufrido un infarto de miocardio en el mes anterior al tratamiento, pacientes con angina de pecho inestable, o un accidente cerebrovascular en el mes anterior al tratamiento, que presenten o que corran el riesgo de trombosis venosa profunda tal como antecedente de enfermedad tromboembólica venosa. RECORMON® Multidosis contiene alcohol bencílico como conservador y, por tanto no debe administrarse a lactantes o niños menores de 3 años.
Precauciones generales: RECORMON® debe utilizarse con precaución en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis o insuficiencia hepática crónica. De igual modo, debe comprobarse que el paciente no sufre una carencia de ácido fólico o vitamina B12, ya que reduciría la eficacia de RECORMON®. La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de RECORMON®. La utilidad de RECORMON® en enfermos nefroscléroticos aún no sometidos a diálisis debe definirse individualmente ya que no puede descartarse con certeza la posible aceleración de la insuficiencia renal. Se ha reportado aplasia pura de células rojas causada por anticuerpos anti-eritropoyetina en pacientes con tratamiento con sustancias eritropoyéticas, incluido RECORMON®. Estos anticuerpos han mostrado reacción cruzada con todas las proteínas eritropoyéticas, y en los pacientes en los que se sospecha o confirma que tienen estos anticuerpos, no deben cambiar a RECORMON® (ver Reacciones adversas). En pacientes con insuficiencia renal durante el tratamiento con RECORMON®, particularmente tras la administración intravenosa, puede producirse un aumento dosis-dependiente del recuento plaquetario, pero que permanece dentro de los límites de la normalidad y regresa a los valores basales en el transcurso de la terapia. No obstante, se recomienda determinar regularmente el recuento de plaquetas durante las 8 primeras semanas de tratamiento. En niños prematuros, deberá realizarse un recuento plaquetario a intervalos regulares, puesto que éste podría estar ligeramente elevado, sobre todo en los primeros 12-14 días de vida. Efecto en el crecimiento de tumores: las eritropoyetinas son factores de crecimiento que estimulan la producción de células rojas. Los receptores de eritropoyetina pueden ser expresados, en la superficie de una variedad de las células tumorales. Como con todos los factores de crecimiento, hay la preocupación de que las eritropoyetinas pueden estimular el crecimiento de cualquier tipo de malignidad. Dos estudios clínicos controlados en los cuales se administró eritropoyetina a pacientes con diferentes cánceres, incluyendo cáncer de cabeza y cuello y cáncer de mama, han mostrado una mortalidad inexplicablemente mayor. En pacientes con cáncer, debe controlarse la cifra plaquetaria y el nivel de hemoglobina a intervalos regulares. En pacientes en programa de autotransfusión, cabe la posibilidad de un aumento del número de plaquetas, por lo general dentro de los límites de la normalidad. Se recomienda, por tanto, determinar el recuento plaquetario al menos una vez por semana. Si este aumento es superior a 150 x 109/l o si la cifra de plaquetas rebasa los márgenes de la normalidad, se retirará la administración de RECORMON®. En pacientes con insuficiencia renal crónica, a menudo hay que aumentar la dosis de heparina durante la hemodiálisis como consecuencia del hematocrito elevado. Si la heparinización no es óptima, cabe la posibilidad de oclusión del sistema de diálisis. Adicionalmente, en pacientes con riesgo de trombosis de la fístula, se recomienda una pronta revisión de ésta, y la profilaxis antitrombótica mediante la administración, por ejemplo, de ácido acetilsalicílico. Los niveles de potasio en suero deben monitorearse frecuentemente durante la terapia con RECORMON®. La elevación de niveles de potasio se ha reportado en algunos pacientes urémicos que recibieron RECORMON®, aunque no se ha establecido la causalidad. En el caso de observar un nivel elevado de potasio, se debe considerar suspender el tratamiento hasta que se normalicen los niveles. Cuando se utilice RECORMON® en una autotransfusión programada, se tendrán en cuenta las directrices oficiales sobre las donaciones sanguíneas, particularmente las siguientes: sólo deben donar sangre los pacientes con un hematocrito ≥33% (hemoglobina ≥11 g/dl [6,83mmol/l]); tener especial cautela si el paciente pesa menos de 50 kg; el volumen de sangre de una extracción no debe superar el 12% del volumen sanguíneo estimado del paciente. El tratamiento debe reservarse para los pacientes en los que se considere de especial importancia evitar las transfusiones de sangre autóloga, una vez evaluada la relación riesgos/beneficios de estas transfusiones. El mal uso del producto por personas sanas (p.e. para dopaje) puede llevar a un aumento excesivo del hematocrito, lo cual puede originar complicaciones cardiovasculares potencialmente letales. RECORMON® en jeringa precargada contiene hasta 0,3 mg de fenilalanina por jeringa y RECORMON® Multidosis contiene 0,5 mg de fenilalanina por vial como excipiente. Por esto, debe usarse con precaución en pacientes afectados por diversas formas de fenilcetonuria. Efectos sobre la capacidad para conducir y utilizar máquinas: RECORMON®no tiene influencia en la habilidad para conducir y utilizar máquinas.
Restricciones de uso durante el embarazo y la lactancia: Los estudios realizados en animales no ha revelado ningún indicio de teratogenicidad de la eritropoyetina beta en esquemas que no conduzcan a un hematocrito excesivamente alto. Ahora bien, dado que no existe suficiente experiencia sobre el uso de RECORMON® en mujeres embarazadas o en lactancia, RECORMON® sólo debe utilizarse durante el embarazo y la lactancia cuando los beneficios esperados justifiquen el riesgo para el feto.
Reacciones secundarias y adversas: Basados en los resultados de ensayos clínicos que incluyen 1.725 pacientes, se estima que aproximadamente el 8% de los pacientes tratados con RECORMON® experimentaron reacciones adversas. Se observaron reacciones adversas durante el tratamiento con RECORMON® predominantemente en pacientes con alteración renal crónica o con tumores malignos subyacentes y lo más frecuente es un aumento de la presión arterial o un agravamiento de hipertensión ya existente y dolor de cabeza. Sistema cardiovascular: pacientes anémicos con insuficiencia renal crónica: la reacción adversa más frecuente en tratamiento con RECORMON® es un aumento de la presión arterial o agravamiento de la hipertensión existente, en especial en casos de aumento rápido del hematocrito. Estas elevaciones de la presión arterial pueden ser tratadas con fármacos. Si no responden al tratamiento farmacológico, se recomienda retirar transitoriamente RECORMON®. Particularmente, al comienzo de la terapia, conviene medir a intervalos regulares la presión arterial, también entre las diálisis. En algunos pacientes con presión arterial normal o baja, pueden sobrevenir crisis hipertensivas con síntomas encefalopatiformes (p. ej.: cefalea, estado de confusión mental, trastornos sensorimotores -como alteraciones del habla o de la marcha- hasta convulsiones tónico-clónicas), lo que requiere atención y cuidados médicos inmediatos. Esto requiere atención médica inmediata y cuidado médico intensivo. Especial atención debe prestarse a las cefaleas súbitas lacerantes de tipo migrañoso como posible señal de advertencia. Pacientes con cáncer: ocasionalmente, puede producirse un aumento de la tensión arterial que puede tratarse con fármacos. Se recomienda, pues, controlar la presión arterial, en particular durante la fase inicial de la terapia. También pueden producirse cefaleas de manera ocasional. Sangre: pacientes anémicos con insuficiencia renal crónica: puede producirse trombosis de la fístula arteriovenosa (shunt), sobre todo en los pacientes con tendencia a la hipotensión o cuya fístula arteriovenosa presente complicaciones (p.ej estenosis y aneurismas), vea Precauciones generales. En la mayoría de los casos, se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del hematocrito. Por esta razón, se recomienda sustituir el hierro por vía oral con una dosis diaria de 200-300 mg Fe2+ en todos los pacientes con valores séricos de ferritina inferiores a 100 mg/l o que presenten una saturación de transferrina por debajo del 20%. En casos aislados, se han observado incrementos transitorios en los niveles séricos de potasio y fosfato, por lo que se recomienda un seguimiento regular de dichos parámetros. En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) asociadas con la terapia con RECORMON®. En caso de ser diagnosticada la APCR, se debe cesar el tratamiento con eritropoyetina y los pacientes no deben cambiar a otra proteína eritropoyética (ver Precauciones generales). Prematuros: en la mayoría de los casos, descienden los valores de ferritinina sérica. Por lo tanto, debe iniciarse lo antes posible (dentro de los primeros 14 días de vida) una terapia oral con 2 mg de hierro (Fe2+) al día. La dosis de hierro debe adaptarse a la concentración sérica de la ferritina. Si esta se halla por debajo de 100 mg/l o si hay otros signos de deficiencia férrica, la administración de Fe2+ deberá aumentarse a 5-10 mg diarios de Fe2+. El tratamiento con hierro debe proseguirse hasta la desaparición de los signos de deficiencia. Pacientes con cáncer: en algunos pacientes, desciende el hierro sérico. Por esta razón, se recomienda sustituir el hierro por vía oral con una dosis diaria de 200-300 mg de hierro en todos los pacientes con valores séricos de ferritina inferiores a 100 mg/l o que presenten una saturación de transferrina por debajo del 20%. En pacientes con mieloma múltiple, linfoma no-Hodgkin o leucemia linfocítica crónica, con saturación de la transferrina inferior al 25%, se han empleado 100 mg de Fe3+ intravenoso por semana. Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es ligeramente más alta en pacientes con cáncer tratados con RECORMON® que en los grupos control tratados o no tratados con placebo. En pacientes tratados con RECORMON®, esta incidencia es 5,9% comparada con el 4,2% de los grupos control; esto no está asociado con un incremento en la mortalidad tromboembólica comparada con los grupos control. Pacientes en un programa de autotransfusión: entre los pacientes sometidos a una autotransfusión programada, se ha observado una incidencia ligeramente mayor de eventos tromboembólicos. Ahora bien, no ha podido establecerse una relación causal con la eritropoyetina beta. Puesto que hay signos de deficiencia transitoria de hierro, todos los pacientes deben recibir por vía oral 300 mg diarios de Fe2+ desde el comienzo del tratamiento con RECORMON® hasta la normalización de los valores de la ferritina. Si a pesar de la sustitución del hierro, se produce una deficiencia de este mineral (valor de la ferritina igual o inferior a 20 mg/l o la saturación de transferrina por debajo del 20%), se considerará la conveniencia de administrar complementariamente hierro por vía intravenosa. Otros: raramente pueden ocurrir reacciones cutáneas como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos aislados se han observado reacciones anafilácticas. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad. Se han comunicado casos aislados de síntomas gripales como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días. A continuación se relacionan las incidencias de reacciones adversas relacionadas con el tratamiento de RECORMON® en ensayos clínicos, clasificados por indicación: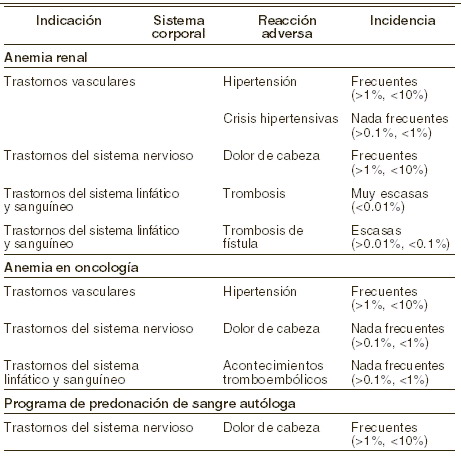
Interacciones medicamentosas y de otro género: Los resultados clínicos obtenidos hasta el momento no ponen en manifiesto interacción alguna de RECORMON® con otros medicamentos. Experimentos con animales revelan que la eritropoyetina beta no incrementa la mielotoxicidad de las drogas citostáticas como la etoposida, cisplatino, ciclofosfamida y fluorouracilo. Incompatibilidades: para evitar problemas de incompatibilidad o pérdida de actividad, no debe mezclarse RECORMON® con otros fármacos o soluciones para infusión. RECORMON®multidosis: para garantizar la conservación del producto durante todo el período de utilización, han de seguirse las siguientes instrucciones: debe emplearse únicamente con el disolvente que le acompaña. No mezclar RECORMON® con otros fármacos. No usar materiales de vidrio para la inyección. Usar exclusivamente materiales de plástico.
Alteraciones en los resultados de pruebas de laboratorio: Las concentraciones séricas de potasio y fosfato han de medirse regularmente durante el tratamiento con RECORMON®. Se ha descrito el aumento de potasio en algunos pacientes urémicos que recibían RECORMON®, pero no se conoce ningún fallecimiento. En caso de cifras altas o crecientes de potasio sérico, se considerará la conveniencia de retirar RECORMON® hasta que se normalice la concentración.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: La experimentación animal no ha revelado ningún indicio de teratogenicidad de la eritropoyetina beta en pautas que no conduzcan a un hematocrito excesivamente alto.
Dosis y vía de administración: La terapia con RECORMON® debe iniciarse por médicos con experiencia en las indicaciones arriba mencionadas. Dado que ha habido algunos casos aislados de reacciones anafilácticas, se recomienda administrar la primera dosis bajo supervisión médica. Las jeringas precargadas de RECORMON® se presentan listas para el uso. Sólo debe administrarse la solución si reúne las condiciones de estar límpida o sólo ligeramente opalescente, ser incolora y contener prácticamente ninguna partícula visible. RECORMON® en jeringas precargadas es un producto estéril pero sin conservadores. En ningún caso debe administrarse más de una dosis por jeringa. La preparación multidosis puede utilizarse en varios pacientes. Para evitar el riesgo de infección cruzada se deben aplicar técnicas asépticas y utilizar siempre, para cada administración, jeringas y agujas estériles desechables. Verifíquese en todo momento, que sólo se tiene en uso un vial de RECORMON® Multidosis (es decir, reconstituido). Tratamiento de pacientes anémicos con insuficiencia renal crónica: la solución reconstituida puede ser administrada por vía subcutánea o intravenosa. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej., en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis. En pacientes no hemodializados, debe preferirse la administración subcutánea a fin de evitar la punción de venas periféricas. El objetivo del tratamiento es incrementar el hematocrito a 30-35% debiendo ser el aumento semanal de por lo menos 0,5%. No debe excederse del valor de 35%. En presencia de hipertensión o de enfermedades cardiovascular, cerebrovascular o vascular periférica, se determinarán individualmente el aumento semanal del hematocrito y el valor deseado de éste, teniendo en cuenta el cuadro clínico del paciente. En ciertos casos, el hematocrito óptimo puede ser inferior a 30%. El tratamiento de la anemia por insuficiencia renal con RECORMON® consta de dos fases: 1.- Fase de corrección: administración subcutánea: la dosis inicial es de 20 UI/kg de peso corporal, tres veces por semana. Si el aumento del hematocrito no ha sido suficiente ( < 0,5 % por semana), esta dosis podrá elevarse cada 4 semanas en otras 20 UI/kg tres veces por semana. La dosis semanal puede fraccionarse en subdosis diarias. Administración intravenosa: la dosis inicial es 40 UI/kg tres veces por semana, Al cabo de 4 semanas, esta dosis puede aumentarse a 80 UI/kg tres veces a la semana. Si es preciso aumentarla de nuevo, deben hacerse a intervalos mensuales a razón de 20 UI/kg de peso corporal tres veces a la semana. La dosis máxima no debe superar 720 UI/kg/semana por ninguna de ambas vías de administración. 2.- Fase de mantenimiento: para mantener el valor hematocrito entre 30 y 35%, primero debe reducirse la dosis a la mitad de la previamente administrada. Después es ajustada individualmente con intervalos de una a dos semanas (dosis de mantenimiento). En caso de administración subcutánea, la dosis semanal puede administrarse con una inyección a la semana o bien dividiendo la dosis en tres o siete veces a la semana. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas, administrando la dosis total requerida en dos semanas. Los resultados de los estudios clínicos en niños revelan que, en general, cuanto menor es la edad, mayor la dosis requerida de RECORMON®. No obstante, debe seguirse la pauta posológica recomendada, puesto que no puede preverse cual será la respuesta individual. Por lo general, el tratamiento con RECORMON® es de larga duración. Sin embargo, en caso de ser necesario puede interrumpirse en cualquier momento. El dato de la dosificación de una vez a la semana está basado en estudios clínicos con un tratamiento de duración de 24 semanas. Prevención de anemia en prematuros: la solución se administra por vía subcutánea en una dosis de 250 UI/kg de peso corporal, tres veces por semana. El tratamiento con RECORMON® debe iniciarse lo antes posible, preferiblemente en los tres primeros días de vida. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con RECORMON® no se beneficien tanto como los prematuros no transfundidos. La terapia debe mantenerse durante 6 semanas. Tratamiento de anemia sintomática en pacientes con cáncer: la solución reconstituida se administra por vía subcutánea, la dosis semanal puede aplicarse en una inyección semanal o en dosis divididas de 3 a 7 veces por semana. La dosis inicial recomendada es 30,000 UI por semana (correspondiente a 450 UI/kg de peso corporal por semana, basado en un peso promedio del paciente). El tratamiento con RECORMON® está indicado si el valor de hemoglobina ≤11 g/dl (6,83mmol/l). El valor de la hemoglobina no debe exceder 13 g/dl (8,07 mmol/l). Si después de 4 semanas de terapia el valor de hemoglobina ha incrementado por lo menos 1 g/dl (0,62 mmol/l), deberá continuar con la dosis actual. Si el valor de hemoglobina no ha incrementado en por lo menos 1 g/dl (0,62 mmol/l), deberá considerarse duplicar la dosis semanal. Si después de 8 semanas de terapia el valor de la hemoglobina no ha incrementado por lo menos 1 g/dl (0,62 mmol/l), la respuesta es poco probable y el tratamiento deberá ser suspendido. La terapia deberá continuar hasta 4 semanas después de terminar la quimioterapia. La dosis máxima no debe exceder 60.000 UI por semana. Una vez que sea logrado el objetivo terapéutico en el paciente, la dosis debe reducirse de 25 a 50% para mantener la hemoglobina a ese nivel. Si se requiere, se puede aplicar una disminución de dosis posterior para asegurar que el nivel de hemoglobina no exceda 13 -g/dl. Si el aumento de hemoglobina es mayor a 2 -g/dl (1,3 -mmol/l) en 4 semanas, la dosis deberá reducirse al 25 a 50%. Tratamiento para incrementar el rendimiento de la sangre autóloga donada: RECORMON® es administrado por vía intravenosa a lo largo de 2 minutos o por vía subcutánea dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir cuando sea ≥33%, RECORMON® es administrado al final de la donación de sangre. Durante la totalidad del período de tratamiento no debe excederse un valor de hematocrito del 48%. La dosis debe ser determinada individualmente para cada paciente por el equipo quirúrgico en función del volumen requerido de sangre predonada y la reserva endógena de eritrocitos: 1.- El volumen necesario de sangre predonada dependerá de la pérdida prevista de sangre, de los medios para conservarla y del estado físico del paciente. Este volumen debería ser suficiente para evitar transfusiones de sangre homóloga. La cantidad requerida de sangre pre-donada se expresa en unidades, equivaliendo una unidad del nomograma a 180 ml de eritrocitos. 2.- La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y el hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la siguiente fórmula: reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33): 100. Hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1.600 (ml). Mujeres: volumen sanguíneo (ml) = 41 (ml/kg)x peso corporal (kg) + 1.200 (ml). (peso corporal ≥45 kg). La indicación para un tratamiento con RECORMON® y, en su caso, la dosis única debe determinarse en función del volumen requerido de sangre predonada y la reserva endógena de eritrocitos, según los gráficos siguientes: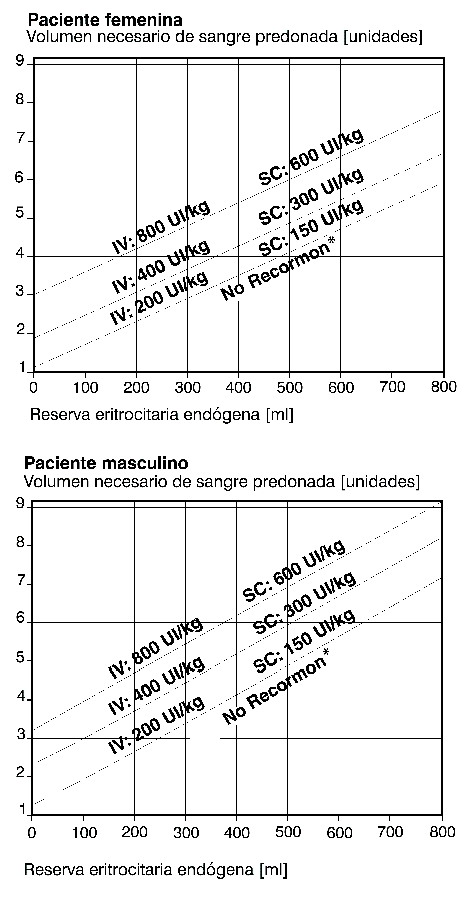
La dosis así determinada se administra dos veces por semana, durante 4 semanas. La dosis máxima no debe exceder de 1.600 UI/kg/semana en administración IV y de 1.200 UI/kg/semana en administración SC. Instrucciones para la manipulación: jeringas precargadas: lávese las manos. Saque una jeringa del envase y asegúrese de que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa. Saque la aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de RECORMON® en la jeringa sea la prescrita. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de RECORMON®. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril. Este medicamento es para aplicarse una sola vez. Cualquier material remanente debe ser desechado. Multidosis: RECORMON® Multidosis se presenta en viales en forma de polvo para solución inyectable. Para disolver el polvo, debe utilizarse la ampolleta de disolvente incluida en el envase, utilizando para ello un dispositivo de reconstitución y extracción como se describe en las instrucciones que siguen. Sólo debe administrarse la solución si reúne las condiciones de estar límpida o sólo ligeramente opalescente, ser incolora y contener prácticamente ninguna partícula visible. No use materiales de vidrio para la inyección, use sólo materiales plásticos. Es esta una preparación multidosis de la que, tras la disolución, pueden extraerse varias dosis a lo largo de un mes. Para evitar el riesgo de que se contamine la solución, se deben aplicar técnicas asépticas (es decir, utilizar jeringas y agujas estériles desechables para administrar cada dosis) y seguir exactamente las instrucciones siguientes. Antes de extraer cada dosis, se desinfectará con alcohol el tapón de caucho del dispositivo, a fin de prevenir la contaminación de la solución en las repetidas perforaciones. Preparación de la solución de RECORMON® Multidosis: saque del estuche el vial con el polvo liofilizado. Anote en la etiqueta las fechas de reconstitución y caducidad (fecha de caducidad: 1 mes después de la reconstitución). Quite el protector de plástico del vial. Desinfecte la tapa de goma con alcohol. Extraiga el dispositivo de reconstitución y de extracción (que permite el intercambio de aire estéril) del blíster protector y retire el capuchón del extremo superior del dispositivo. Coloque el dispositivo en el vial hasta que el cierre se coloque en su sitio (deberá oirse la señal de ensamblaje del resorte). Coloque la aguja verde en la jeringa (contenidas ambas en el estuche) y quite el capuchón protector de la aguja. Mantenga la ampolleta con el punto azul hacia arriba. Agite o golpee ligeramente la ampolleta para que el líquido que esté en el cuello de la misma baje al cuerpo. Sujete el cuello de la ampolleta entre los dedos y rómpalo en dirección opuesta a usted. Transfiera todo el disolvente hacia la jeringa. Desinfecte el tapón de goma con alcohol. Perfore el tapón con la aguja hasta una profundidad de 1 cm, aproximadamente, e inyecte poco a poco todo el disolvente en el vial. Después retire la jeringa (con la aguja) del dispositivo. Gire el vial suavemente hasta que el polvo se haya disuelto. No agite. Compruebe que la disolución es transparente y prácticamente libre de partículas. Ponga el tapón protector encima del dispositivo. Antes y después de la reconstitución, RECORMON® Multidosis debe conservarse a una temperatura de 2-8°C (refrigerador). Preparación de una inyección única: antes de extraer cada dosis, retire el capuchón y desinfecte con alcohol el tapón de goma del dispositivo a utilizar. Coloque una aguja de 26G dentro de la jeringa desechable adecuada (máx. 1 ml). Retire el capuchón protector de la aguja e insértela en el tapón de goma del dispositivo. Extraiga con la jeringa la solución de RECORMON®, saque el aire de la jeringa al vial y ajuste la cantidad de solución de RECORMON® en la jeringa a la dosis prescrita. Después retire la jeringa (con aguja) del dispositivo. Sustituya la aguja por una nueva (la nueva aguja debe ser del tamaño que suele utilizarse para las inyecciones) Retire el capuchón protector y expulse cuidadosamente el aire de la aguja sosteniendo la jeringa en posición vertical y presionando suavemente el émbolo hacia arriba, hasta que aparezca una gota de líquido en la punta de la aguja. Para una inyección subcutánea limpie la piel en el sitio de inyección con un algodón empapado en alcohol. Forme un pliegue de piel pellizcando la piel con los dedos pulgar e índice. Sujete la jeringa por la parte más próxima a la aguja e inserte la aguja en la piel (en el pliegue) con un movimiento firme y rápido. Inyecte la solución de RECORMON®. Retírese la aguja rápidamente y haga presión sobre el lugar de inyección con una gasa seca y estéril.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: El margen terapéutico de RECORMON® es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
Presentación(es): Caja con 1 o 6 jeringas precargadas con 5.000 UI de eritropoyetina beta en 0,3 ml. Caja con 1 frasco ámpula multidosis con liofilizado con 50.000 UI de eritropoyetina beta, ampolleta con 10 ml de diluyente y un dispositivo que permite el intercambio de aire estéril.
Recomendaciones sobre almacenamiento: No se agite. No se congele. Protéjase de la luz. Consérvese en refrigeración, de 2 a 8°C dentro de su empaque original. RECORMON® en jeringa precargada es estable fuera del refrigerador, a temperatura ambiente a no más de 25°C durante un máximo de 3 días seguidos, sólo por una ocasión. RECORMON® en frasco ámpula multidosis con liofilizado es estable fuera del refrigerador, a temperatura ambiente a no más de 25°C durante un máximo de 5 días seguidos sólo por una ocasión. Hecha la mezcla, el producto se conserva estable durante 1 mes en refrigeración de 2 a 8°C.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños.
Nombre y domicilio del laboratorio: Hecho en Alemania por: Roche Diagnostics GmbH, Sandhofer Strasse 116, 68305, Mannheim, Alemania. Acondicionado y distribuido por: Productos Roche S.A. de C.V. Vía I. Fabela Nte. No. 1536-B, 50030, Toluca, México. Para información bibliográfica, comuníquese a nuestro centro de información médica, Tel. (01) (55) 52585099 y 01-800-0076243, o mexico.info@roche.com. ®Marca registrada.
Número de registro del medicamento: 529M95 SSA IV.
Clave de IPPA: HEAR-083501CT050039/RM 2008