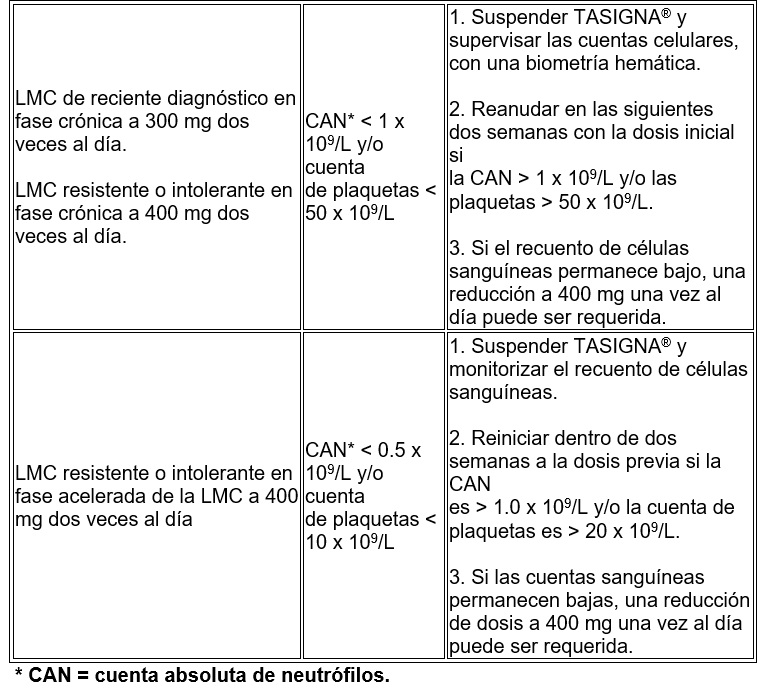
TASIGNA
CARNOT
Denominación genérica: Nilotinib.
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: Clorhidrato monohidratado de nilotinib equivalente a 200 o 150 mg de nilotinib Excipiente, c.b.p. 1 cápsula.
Indicaciones terapéuticas: TASIGNA® está indicado para el tratamiento de la fase crónica (FC) y la fase acelerada (FA) de la leucemia mieloide crónica (LMC), positiva para el cromosoma Filadelfia, en pacientes adultos resistentes o intolerantes a por lo menos un tratamiento previo incluyendo el imatinib. Tratamiento de pacientes adultos con leucemia mieloide crónica con cromosoma Filadelfia positivo (LMC Ph+) de reciente diagnóstico en fase crónica.
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Absorción: Las concentraciones pico de nilotinib se logran 3 horas después de su administración oral. La absorción de nilotinib posterior a la administración oral fue de aproximadamente 30%. En voluntarios sanos, el Cmáx. y el área debajo de la curva de la concentración sérica (ABC) de nilotinib aumentaron en 112 y 82%, respectivamente en comparación con las condiciones de ayuno cuando se administró TASIGNA® con los alimentos. La administración de TASIGNA® 30 minutos o 2 horas después de los alimentos aumentó la biodisponibilidad de nilotinib en 29 o 15%, respectivamente (véase Dosis y vía de administración, Precauciones generales e Interacciones medicamentosas y de otro género). Distribución: La relación sangre-plasma de nilotinib es de 0.68. El enlace proteico-plasmático es de aproximadamente 98% con base en los experimentos realizados in vitro. Biotransformación: Las vías metabólicas principales identificadas en sujetos sanos son la oxidación y la hidroxilación. El nilotinib es el componente de mayor circulación en el suero. Ninguno de los metabolitos contribuye significativamente a la actividad farmacológica de nilotinib. Eliminación: Después de una dosis única de nilotinib radiomarcado en sujetos sanos, más de 90% de la dosis fue eliminada en 7 días, principalmente en las heces. El fármaco madre constituyó 69% de la dosis. Linearidad/no-linearidad: La exposición a nilotinib en estado estable fue dependiente de la dosis, con aumentos menores proporcionales a la dosis en la exposición sistémica a niveles de dosis mayores a 400 mg administrados como la dosificación de una vez al día. La exposición sérica diaria de nilotinib de 400 mg dos veces al día en estado estable fue 35% mayor que la dosificación de 800 mg una vez al día. La exposición sistémica (ABC) de nilotinib en estado en equilibrio a un nivel de dosis de 400 mg dos veces al día fue de aproximadamente 13.4% más alta que con 300 mg dos veces al día. El promedio de las concentraciones pico y mínima de nilotinib 12 meses fueron aproximadamente 15.7 y 14.8% más altas después de la dosis de 400 mg dos veces al día en comparación con 300 mg dos veces al día. No hubo un aumento relevante de exposición al nilotinib cuando la dosis aumentó de 400 a 600 mg dos veces al día. Características en pacientes: Las condiciones de estado estable fueron logradas para el octavo día. Un aumento en la exposición sérica de nilotinib entre la primera dosis y el estado estable fue de aproximadamente dos veces para la dosis diaria y de 3.8 veces para la dosificación de dos veces al día. La vida media de eliminación aparente estimada de la dosis múltiple farmacocinética con la dosis diaria fue de aproximadamente 17 horas. La variabilidad interpaciente de la farmacocinética de nilotinib fue de moderado a alto. Propiedades farmacodinámicas: Grupo farmacoterapéutico: Agente antineoplásico - inhibidor de la proteína tirosin-cinasa. Código ATC: bajo asignación. TASIGNA® es un inhibidor potente y selectivo de la actividad de la tirosin-cinasa Abl y de la oncoproteína Bcr-Abl, ambas en líneas celulares y en las células leucémicas positivas para el cromosoma Filadelfia. El fármaco se fija fuertemente dentro del sitio de fijación del ATP de tal manera que sirve como un inhibidor potente del tipo salvaje del Bcr-Abl y mantiene su actividad contra 32/33 formas mutantes imatinib-resistente del Bcr-Abl. Como consecuencia de esta actividad bioquímica, el nilotinib inhibe selectivamente la proliferación e induce la apoptosis en las líneas celulares y en las células leucémicas primarias positivas del cromosoma Filadelfia de pacientes LMC. En modelos murinos de LMC, como agente único, el nilotinib disminuye la carga tumoral y prolonga la supervivencia posterior a su ingesta oral. TASIGNA® tiene poco o ningún efecto contra la mayoría de las demás cinasas estudiadas, incluyendo el Src, excepto por el PDGF, Kit CSF-1R, DDR y cinasas del receptor de Ephrin el cual inhibe a concentraciones dentro del rango logrado posterior a la administración oral de dosis terapéuticas recomendadas para el tratamiento del LMC (véase tabla 1).
Tabla 1. Perfil Cinasa del Nilotinib (Fosforilación IC 50 nM).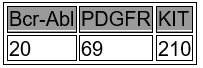
Estudios clínicos: LMC Ph+ en fase crónica (FC) de reciente diagnóstico. Un estudio clínico abierto, multicéntrico, aleatorizado de Fase III fue realizado para determinar la eficacia de TASIGNA® vs. Glivec en pacientes adultos con LMCPh+ en fase crónica de reciente diagnóstico y confirmada citogenéticamente. Los pacientes se encontraban dentro de los primeros seis meses desde el diagnóstico y no fueron tratados previamente para LMC-FC, excepto por hidroxiurea y/o anagrelida. Además, los pacientes fueron estratificados de acuerdo con el valor de riesgo Sokal al momento del diagnóstico. La eficacia se basó en un total de 846 pacientes (283 pacientes en el grupo de imatinib 400 mg una vez al día, 282 pacientes en el grupo de nilotinib 300 mg dos veces al día, 281 pacientes en el grupo de nilotinib 400 mg dos veces al día). Las características basales estuvieron bien balanceadas entre los tres grupos. La mediana de edad fue de 46 años en el grupo de imatinib y 47 años en ambos grupos de nilotinib, con 12.4, 12.8 y 10.0% fue ≥ 65 años de edad en los grupos de tratamiento de imatinib, nilotinib 300 mg dos veces al día y nilotinib 400 mg dos veces al día, respectivamente). Más de 60% de todos los pacientes eran caucásicos y 25% eran asiáticos. El punto de tiempo de análisis de datos primarios se realizó cuando los 846 pacientes completaron 12 meses de tratamiento (o descontinuaron antes). La mediana de tiempo del tratamiento fue de 14 meses en los tres grupos de tratamiento. En cada brazo de tratamiento, más de 60% de los pacientes habían recibido tratamiento por más de 12 meses. La mediana de la intensidad de la dosis actual fue de 400 mg/día en el grupo de imatinib, 592.2 mg/día en el grupo de nilotinib 300 mg dos veces al día y 778.8 mg/día en el grupo de nilotinib 400 mg dos veces al día. Este estudio continua. Respuesta Molecular Mayor (RMM): La variable primaria de eficacia fue la RMM a los 12 meses después de iniciar el medicamento de estudio. La RMM se definió como un valor de porcentaje de ≤ 0.1% BCR-ABL/ABL medido mediante la escala internacional por RQ-PCR, lo cual corresponde a ≥ 3 de reducción log de los transcritos de BCR-ABL a partir de una línea basal estandarizada. La variable de eficacia primaria es decir la tasa de Respuesta Molecular Mayor (RMM) a los 12 meses fue estadísticamente significativa más alta en el grupo de nilotinib 300 mg dos veces al día en comparación con el grupo de imatinib 400 mg una vez al día (44.3 vs. 22.3%, p < 0.0001). La tasa de Respuesta Molecular Mayor (RMM) a los 12 meses, también fue estadísticamente significativa, más alta en el grupo de nilotinib dos veces al día 400 mg en comparación con el grupo de imatinib 400 mg una vez al día (42.7 vs. 22.3%, p < 0.0001) (véase tabla 2). Con la dosis recomendada de nilotinib de 300 mg dos veces al día, la tasa de RMM a los 3, 6, 9 y 12 meses fueron 8.9, 33.0, 43.3 y 44.3%. En el grupo de nilotinib 400 mg dos veces al día, el índice de RMM a los 3, 6, 9 y 12 meses fueron 5.0, 29.5, 38.1 y 42.7%. En el grupo de imatinib 400 mg una vez al día, el índice de RMM a los 3, 6, 9 y 12 meses fueron 0.7, 12.0, 18.0 y 22.3%. Los análisis Kaplan-Meier del tiempo para obtener la primera RMM entre todos los pacientes se muestran gráficamente en la figura 1. La probabilidad de alcanzar RMM en diferentes puntos de tiempo fue mayor en ambos grupos de nilotinib en comparación con el grupo de imatinib (HR = 2.6 y log-rank estratificado p < 0.0001 entre nilotinib 300 mg dos veces al día e imatinib, HR = 1.6 y log-rank estratificado p < 0.0001 entre nilotinib 400 mg dos veces al día e imatinib). Las proporciones de pacientes que tuvieron relaciones de BCR-ABL que cayeron en las categorías ≤ 0.01% (reducción de 4 logaritmos) y ≤ 0.0032% (reducción de 4.5 logaritmos) a los 12 meses fueron más elevadas, y fueron estadísticamente significativas en ambos grupos de nilotinib (11.7/4.3% y 8.5/4.6%, respectivamente) en comparación con el grupo de imatinib (3.9/0.4%). Para todos los grupos de riesgo Sokal, los índices de respuesta fueron más altos en los grupos de nilotinib que en el grupo de imatinib.
Tabla 2. Tasas de RMM a los 12 meses.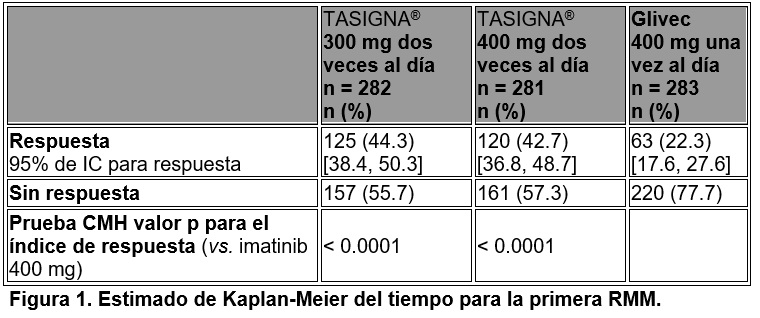
Respuesta citogenética Completa (RCC): La RCC se definió como 0% de metafases Ph + en la médula ósea con base en un mínimo de 20 metafases evaluadas. La mejor tasa de RCC a los 12 meses (incluye los pacientes que alcanzaron la RCC en o antes del punto de tiempo de 12 meses como respondedores) fue estadísticamente mayor para ambos grupos con nilotinib, 300 y 400 mg dos veces al día, en comparación con el grupo de imatinib 400 mg una vez al día (véase tabla 3).
Tabla 3. Mejor tasa de RCC a los 12 meses.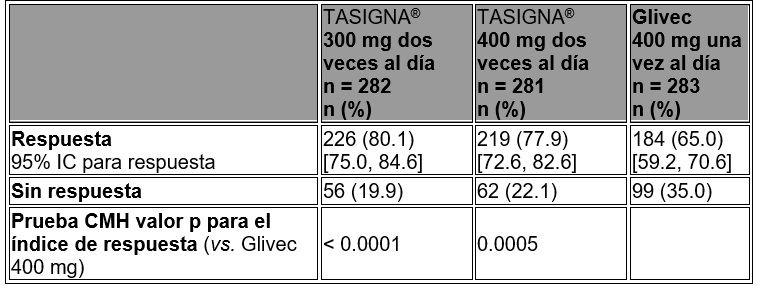
Progresión a Fase Acelerada (FA)/Crisis Blástica (CB) en el tratamiento: Durante el tratamiento, 14 pacientes progresaron a FA o CB (2 en el grupo de nilotinib 300 mg dos veces al día, 1 en el grupo de nilotinib 400 mg dos veces al día y 11 en el grupo de imatinib 400 mg una vez al día). Ningún paciente que progresó había alcanzado una RMM. Sin embargo, 3 pacientes habían alcanzado la RCC (todos en el grupo de imatinib). Las tasas estimadas de pacientes libres de progresión a FA o CB a los 12 meses fueron 99.3, 99.6 y 96.5%, respectivamente. Hubo una diferencia estadísticamente significativa en la tasa de progresión a FA o CB entre nilotinib 300 mg dos veces al día e imatinib (p = 0.0095) y entre nilotinib 400 mg dos veces al día e imatinib (p = 0.0037). LMC Ph+ en fase crónica y LMC Ph+ en fase acelerada resistente o intolerante a imatinib: Un estudio abierto multicéntrico de Fase II fue realizado para determinar la eficacia de TASIGNA® en pacientes con LMC resistentes o intolerantes a imatinib, con brazos separados de tratamiento para la fase crónica y acelerada de la enfermedad. El estudio se está llevando a cabo actualmente. La eficacia se basó en 280 pacientes en fase crónica (FC) y 96 pacientes en fase acelerada (FA) incluidos en el estudio. La duración mediana del tratamiento fue de 261 y 191 días, respectivamente (véase tabla 4). TASIGNA® fue administrado de manera continua (dos veces al día, 2 horas después de los alimentos y sin alimentos adicionales por lo menos durante una hora) a menos que hubiera evidencia de una respuesta inadecuada o la progresión de la enfermedad. Se permitió el aumento de la dosis a 600 mg dos veces al día.
Tabla 4. Duración de la exposición a TASIGNA®
La resistencia a imatinib incluyó la falla para lograr una respuesta hematológica completa (por 3 meses), respuesta citogenética (a los 6 meses) o una respuesta citogenética mayor (a los 12 meses) o una progresión de la enfermedad después de alcanzar una respuesta citogenética o hematológica previa. La intolerancia a imatinib incluyó pacientes que descontinuaron imatinib debido a la toxicidad y no alcanzaron una respuesta citogenética mayor al momento de su inclusión en el estudio. En general, 73% de los pacientes fueron resistentes a imatinib, mientras que 27% fueron tolerantes al mismo. La mayoría de los pacientes tuvieron una larga historia de LMC que incluyó el tratamiento previo extenso con otros agentes antineoplásicos, como el imatinib, hidroxiurea, interferón, y algunos otros que fallaron al trasplante de células madre (véase tabla 5). La dosis mediana previa más alta de imatinib había sido 600 mg/día para los pacientes en fase crónica (CP) y 800 mg/día para los pacientes con fase acelerada (AP), y la dosis previa más alta de imatinib fue > 600 mg/día en 76% de todos los pacientes con 45% de los pacientes recibiendo la dosis de imatinib > 800 mg/día.
Tabla 5. Características de la enfermedad (LMC) en los pacientes en los estudios Fase II.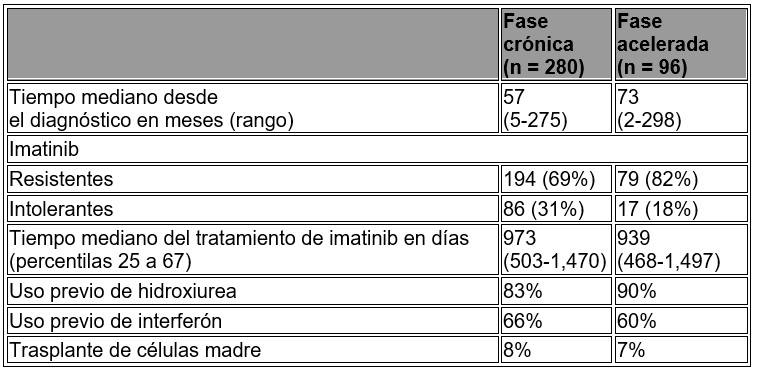
El objetivo primario en los pacientes con LMC en fase crónica fue una respuesta citogenética mayor (RCigM), definida como la eliminación (RCC, respuesta citogenética completa) o una disminución significativa de < 35% metafases Ph+ (respuesta citogenética parcial) de células hematopoyéticas Ph+. La respuesta hematológica completa (RHC) en pacientes CP fue considerado como un punto secundario. El punto primario en los pacientes en fase acelerada (FA) fue una respuesta hematológica confirmada global (RH), definida como una respuesta hematológica completa, ausencia de leucemia o una recaída a la fase crónica. La tasa de RCigM en 280 pacientes en fase crónica fue de 52%. La mayoría logró su RCigM rápidamente en 3 meses (con una mediana de 2.8 meses) de haber iniciado el tratamiento con TASIGNA® y de manera sostenida (la duración mediana no ha sido alcanzada). Los pacientes con una RHC basal lograron una RCigM con mayor rapidez (1 vs. 2.8 meses). De los pacientes en fase crónica sin una RHC basal, 74% logró una RHC y una mediana de tiempo para la RHC de un mes y la mediana de duración de la RHC no ha sido lograda. La tasa global de RH confirmada en 96 pacientes con fase acelerada (AP) fue de 45%. La mayoría logró una RH tempranamente con el tratamiento de TASIGNA® (mediana de 1 mes), que ha sido duradera (la duración mediana no ha sido alcanzada). Ningún paciente ha perdido su estado de RHC. La tasa de RCigM fue de 30% con una mediana de tiempo de respuesta de 2.0 meses. Las tasas de respuesta para los dos brazos de tratamiento se reportan en la tabla 6.
Tabla 6. Respuesta en LMC.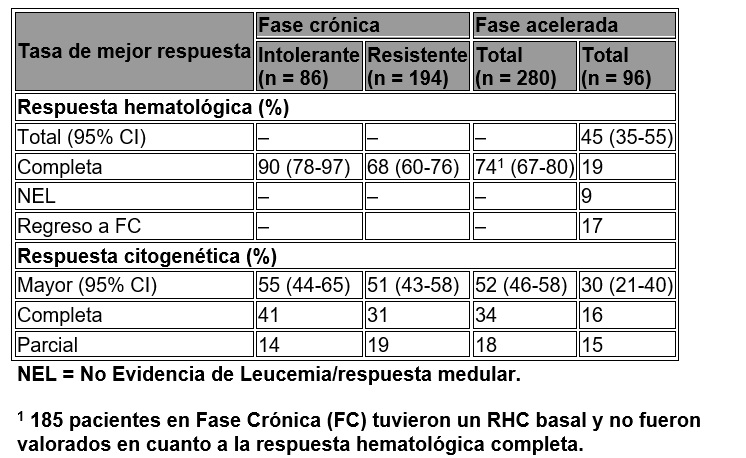
También se incluyeron brazos separados de tratamiento en el estudio de Fase II para estudiar TASIGNA® en un grupo de pacientes con fase crónica (FC) y fase acelerada (FA) que habían sido extensamente pretratados con terapias múltiples incluyendo un agente inhibidor de la tirosin-cinasa además del imatinib. De estos pacientes, 30/36 (83%) eran resistentes al tratamiento, no intolerantes. En 22 pacientes con fase crónica (FC), evaluados para eficacia, TASIGNA® indujo una tasa de RCigM de 32% y una tasa de RHC de 50%. En 11 pacientes con fase acelerada (FA) evaluados para la eficacia, el tratamiento indujo una tasa RH global de 36%. Después de la falta de respuesta con imatinib, 24 mutaciones diferentes de Bcr-Abl fueron notadas en 45% de los pacientes con LMC en fase crónica (FC) y 57% de los pacientes con LMC en fase acelerada (FA) quienes fueron evaluados para mutaciones. TASIGNA® demostró eficacia en pacientes con una variedad de mutaciones Bcr-Abl asociadas con resistencia a imatinib, excepto la mutación T315I.
Contraindicaciones: Hipersensibilidad conocida al nilotinib o a cualquiera de sus excipientes.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: No existen datos sobre el uso de TASIGNA® en mujeres embarazadas. Los estudios realizados en animales no mostraron toxicidad, pero sí se observó embrio y fetotoxicidad con aquellas dosis que también causaron toxicidad materna (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). TASIGNA® no debe ser utilizada durante el embarazo a menos que sea necesario. Si el fármaco es utilizado durante el embarazo, la paciente debe ser informada de los riesgos potenciales al feto. Mujeres en edad reproductiva: Aquellas mujeres en edad reproductiva deben ser aconsejadas en utilizar un método anticonceptivo efectivo durante el tratamiento con TASIGNA®. Lactancia: Se desconoce si el nilotinib es excretado en la leche materna humana. Algunos estudios realizados en animales mostraron que es excretado en la leche materna. Las mujeres no deberían dar de amamantar mientras toman TASIGNA® ya que un riesgo para el infante no puede ser excluido. Fertilidad: No se observaron efectos en la cuenta y la motilidad de las células espermáticas y en la fertilidad de las ratas macho y hembra hasta la dosis elevada más alta de aproximadamente 5 veces más que la dosis recomendada por humanos (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad)
Reacciones secundarias y adversas: Los datos descritos a continuación reflejan la exposición de un total de 717 pacientes a TASIGNA® a partir de un estudio aleatorizado fase III en pacientes recién diagnosticados con LMC Ph+ en fase crónica tratados con la dosis recomendada de 300 mg dos veces al día (n = 279) y a partir de un estudio abierto multi-centro Fase II en pacientes con LMC resistente o intolerante a imatinib en fase crónica (n = 318) y fase acelerada (n = 120) tratados con la dosis recomendada de 400 mg dos veces al día. LMC Ph+ en fase crónica recién diagnosticada: Los datos descritos a continuación reflejan la exposición a TASIGNA® a partir de un estudio aleatorizado fase III en pacientes recién diagnosticados con LMC Ph+ en fase crónica tratados en la dosis recomendada de 300 mg dos veces al día (n = 279). En este estudio, 64% de los pacientes tratados con nilotinib 300 mg dos veces al día tuvieron duraciones de exposición de más de 12 meses y 16.5% de los pacientes tuvieron duraciones de exposición de más de 18 meses. La mediana de tiempo sobre el tratamiento fue de 14 meses. Las Reacciones Adversas al Medicamento (RAM) no hematológicas más frecuentes fueron salpullido, prurito, dolor de cabeza, náuseas, fatiga y mialgia. La mayoría de estos RAMs fueron de severidad media a moderada (Grado 1 o 2). Se observó menos comúnmente dolor abdominal superior, alopecia, constipación, diarrea, piel seca, espasmos musculares, artralgia, dolor abdominal, edema periférico y vómito (≤ 10 y > 5%) y fueron de severidad leve a moderada, manejables y en general no requirieron de la reducción de dosis. Los derrames pleurales y pericárdicos ocurrieron en < 1% de los pacientes que recibieron TASIGNA®. Se reportaron hemorragias gastrointestinales en < 1% de los pacientes. El cambio del nivel basal en la media de tiempo del intervalo QTcF medio promediado en estado en equilibrio con la dosis recomendada de nilotinib de 300 mg dos veces al día fue de 6 mseg. En el grupo de nilotinib 400 mg dos veces al día y el grupo de imatinib 400 mg una vez al día, el intervalo QTcF medio promediado con el tiempo en estado en equilibro fueron de 6 y 3 mseg, respectivamente. Ningún paciente tuvo un QTcF absoluto de > 500 mseg en ninguno de los grupos de tratamiento y no se observaron eventos de torsade de pointes. Un incremento en QTcF desde la base que excediera más de 60 mseg se observó en 3 pacientes (uno en el grupo de tratamiento de 300 mg dos veces al día y dos en el grupo de tratamiento de 400 mg dos veces al día). No se observó disminución en la media de la Fracción de Eyección del Ventrículo Izquierdo (FEVI) desde el nivel basal en ningún momento durante el tratamiento y en ningún grupo de tratamiento. Tampoco hubieron pacientes en ninguno de los grupos de tratamiento con una FEVI de < 45% durante el tratamiento y ningún paciente tuvo una reducción absoluta en FEVI de más de 15%. No se han reportado muertes repentinas. Las RAMs hematológicos incluyen mielosupresión: trombocitopenia (17%), neutropenia (14%) y anemia (6%), (véase tabla 3 para las anomalías de laboratorio grado ¾. Se observó la discontinuación por eventos adversos sin importar la causalidad en 6.8% de los pacientes. LMC Ph+ en fase crónica y LMC Ph+ en fase acelerada resistente o intolerante: Los datos descritos enseguida reflejan la exposición a TASIGNA® en 438 pacientes LMC Ph+ en fase crónica y en fase acelerada resistente o intolerante, al menos a una terapia previa incluyendo imatinib en un estudio multicéntrico abierto. Al corte, 46% de los pacientes con LMC en fase crónica tuvieron duraciones de exposición de 6 a 12 meses y 18% de los pacientes tuvieron una duración de exposición de más de 12 meses, 62% de los pacientes con LMC en fase acelerada tuvieron duraciones de exposición de 3 a 12 meses y 10% de los pacientes tuvieron una duración de exposición de más de 12 meses. La dosis utilizada fue de 400 mg dos veces al día. La duración mediana de la exposición en días para estos pacientes con LMC en fase crónica y fase acelerada fue de 245 (1 a 502) y 138 (2 a 503), respectivamente. Las RAMs más frecuentes ( > 10% en las poblaciones combinadas de pacientes de LMC-FC y LMC-FA) no hematológicos fueron salpullido, prurito, náuseas, fatiga, dolor de cabeza, constipación, y diarrea. La mayoría de estos RAMs fueron de severidad leve a moderada. El dolor de huesos, artralgia, espasmos musculares y edema periférico fueron observados en menor cantidad (≤ 10 y 5%) y fueron de severidad leve a moderada (grado 1 o 2). Los derrames pleurales o pericárdicos así como las complicaciones de retención de líquidos ocurrieron en < 1% de los pacientes que recibieron TASIGNA®. Se observó insuficiencia cardiaca congestiva en < 1% de los pacientes. Se reportó hemorragia gastrointestinal y SNC en 1 y < 1% de los pacientes, respectivamente. Se observó que el intervalo QTcF excedía de 500 mseg en 3 pacientes ( < 1%). No se observaron episodios de torsade de pointes (transitorio o sostenido). Los RAMs hematológicos incluyen mielosupresión: trombocitopenia (27%), neutropenia (15%), y anemia (13%), (véase tabla 6 para las anomalías de laboratorio grado ¾. La suspensión de los eventos adversos, independientemente de la causa, fue observada en 16% de los pacientes en fase crónica y en 14% de los pacientes con fase acelerada. En la tabla 7 se muestran las reacciones adversas al medicamento no hematológicas (excluyendo las anomalías de laboratorio) que se reportaron en al menos 5% de los pacientes de los estudios clínicos con TASIGNA®. Éstas se colocaron en una frecuencia descendente, primero la más frecuente, utilizando la siguiente convención: muy común (≥ 1/10) o común (≥ 1/100, ≤ 1/10). La frecuencia se basa en lo más alto de cualquiera de los grupos de TASIGNA en los dos estudios.
Tabla 7. Reacciones adversas al medicamento no hematológicas más frecuentemente reportadas ( > 5% en cualquier grupo de TASIGNA®)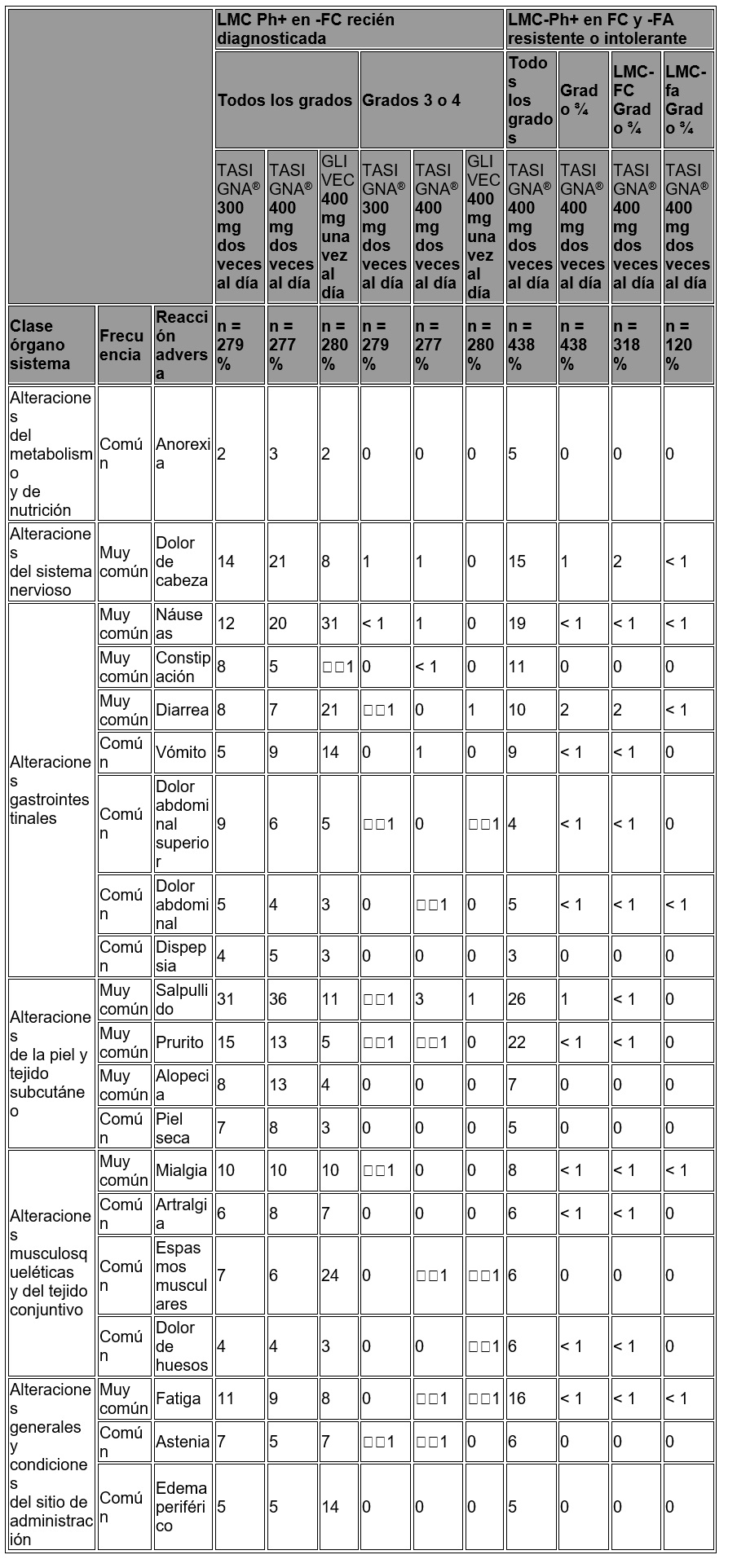
Datos adicionales de los estudios clínicos: Se reportaron las siguientes reacciones adversas al medicamento en pacientes de los estudios clínicos de TASIGNA® en las dosis recomendadas a una frecuencia de menos de 5% (común es ≥ 1/100 y < 1/10; no común es > 1/1,000 y < 1/100; los eventos individuales se capturaron como Frecuencia desconocida). Para las reacciones adversas al medicamento enlistadas bajo "Investigaciones", los eventos muy comunes (≥ 1/10) que no se incluyeron en la tabla 2, también fueron reportados. Estas reacciones adversas se incluyeron con base en la relevancia clínica y se catalogaron en orden decreciente de seriedad dentro de cada categoría. Infecciones e infestaciones: Común: foliculitis. No común: infección del tracto respiratorio superior, neumonía, infección del tracto urinario, gastroenteritis, faringitis. Frecuencia desconocida: sepsis, bronquitis, infección por virus de herpes, candidiasis, absceso subcutáneo, absceso anal, infección del tracto respiratorio, nasofaringitis, rinitis, furúnculo, tinea pedis. Neoplasmas benignos, malignos y no especificados: Común: papiloma de piel. Frecuencia desconocida: papiloma. Alteraciones de la sangre y del sistema linfático: Común: neutropenia febril, linfopenia, pancitopenia. Frecuencia desconocida: trombocitopenia, leucocitosis. Alteraciones del sistema inmune: Frecuencia desconocida: hipersensibilidad. Alteraciones endocrinas: No común: hipertiroidismo. Frecuencia desconocida: hipotiroidismo, hiperparatiroidismo secundario, tiroiditis. Alteraciones del metabolismo y de nutrición: Común: hipocaliemia, diabetes mellitus, hipercolesterolemia, hiperlipidemia, hipomagnasemia, hipercalemia, hiperglucemia. No común: hiponatremia, hipocalcemia, hipofosfatemia, deshidratación, apetito disminuido, apetito incrementado. Frecuencia desconocida: diabetes mellitus, hipercalcemia, hiperfosfatemia, hiperuricemia, gota, hipoglucemia, dislipidemia. Alteraciones psiquiátricas: Común: insomnio. No común: depresión, ansiedad. Frecuencia desconocida: desorientación, estado de confusión, amnesia, disforia. Alteraciones del sistema nervioso: Común: mareo, hipoestesia, parestesia. No común: hemorragia intracraneal, migraña, temblor, hiperestesia. Frecuencia desconocida: edema cerebral, neuritis óptica, neuropatía periférica, síncope, letargia, disestesia. Alteraciones oculares: Común: prurito ocular, conjuntivitis, ojo seco. No común: hemorragia ocular, reducción de agudeza visual, edema periorbital, edema de párpado, fotopsia, conjuntivitis, irritación de ojos. Frecuencia desconocida: papiledema, diplopía, visión borrosa, fotofobia, hinchazón de ojos, blefaritis, dolor de ojos, coriorretinopatía, hemorragia conjuntival, conjuntivitis alérgica, hiperemia conjuntival, hiperemia ocular, enfermedad ocular superficial, hiperemia escleral. Alteraciones del oído y el laberinto: Común: vértigo. Frecuencia desconocida: daño del oído, dolor del oído. Alteraciones cardiacas: Común: palpitaciones, electrocardiograma QT prolongado. No común: falla cardiaca, angina de pecho, fibrilación atrial, derrame pericárdico, enfermedad de arteria coronaria, cianosis, cardiomegalia, murmullo cardiaco, bradicardia. Frecuencia desconocida: infarto al miocardio, disfunción ventricular, pericarditis, latidos cardiacos fuertes, extrasístoles, arritmia, disminución de la fracción de eyección. Alteraciones vasculares: Común: hipertensión, rubor. No común: crisis hipertensa, hematoma. Frecuencia desconocida: choque hemorrágico, hipotensión, trombosis. Alteraciones respiratorias, torácicas y mediastinales: Común: disnea, disnea de esfuerzo, tos, disfonía. No común: edema pulmonar, derrame pleural, enfermedad intersticial pulmonar, dolor pleurítico, pleuresía, epistaxis, dolor faringolaríngeo, irritación de garganta. Frecuencia desconocida: hipertensión pulmonar. Alteraciones gastrointestinales: Común: malestar abdominal, distensión abdominal, flatulencia.No común: pancreatitis, hemorragia gastrointestinal, melena, ulceración de boca, reflujo gastroesofágico, estomatitis, dolor esofágico, disgeusia, boca seca. Frecuencia desconocida: perforación de úlcera gastrointestinal, hemorragia retroperitoneal, hematemesis, úlcera gástrica, esofagitis ulcerativa, oclusión intestinal, gastritis, hemorroides, hernia hiatal, hemorragia rectal, sensibilidad de dientes, gingivitis.Alteraciones hepatobiliares: Común: función hepática anormal. No común: hepatitis, ictericia. Frecuencia desconocida: hepatotoxicidad. Alteraciones de la piel y del tejido subcutáneo: Común: sudoración nocturna, eccema, urticaria, eritema, hiperhidrosis, contusión, acné, dermatitis. No común: salpullido exfoliativo, erupción medicamentosa, dolor de piel, equimosis, cara hinchada. Frecuencia desconocida: eritema nodoso, úlcera de piel, petequias, fotosensibilidad, ampollas, quiste dérmico, hiperplasia sebácea, atrofia de piel, decoloración de piel, exfoliación de piel, hiperpigmentación de piel, hipertrofia de piel. Alteraciones musculosqueléticas y de tejido conjuntivo: Común: dolor musculosquelético de pecho, dolor de espalda, dolor musculosquelético. No común: dolor, debilidad muscular. Frecuencia desconocida: artritis, hinchazón de articulaciones, dolor de costado. Alteraciones renales y urinarias: No común: disuria, micción de urgencia, nocturia, polaquiuria. Frecuencia desconocida: falla renal, hematuria, incontinencia urinaria, cromaturia. Alteraciones del sistema reproductivo y del seno: No común: dolor del seno, ginecomastia, disfunción eréctil. Frecuencia desconocida: endurecimiento del seno, menorragia, hinchazón de pezón. Alteraciones generales y condiciones del sitio de administración: Común: pirexia, dolor de pecho, malestar del pecho. No común: edema facial, edema gravitacional, enfermedad tipo influenza, escalofríos, malestar. Frecuencia desconocida: sensación de calor, edema localizado. Investigaciones: Muy común: incremento de lipasa. Común: disminución de cuenta de plaquetas, incremento de amilasa en sangre, incremento de alanina aminotransferasa, incremento de aspartato aminotransferasa, incremento de bilirrubina en sangre, incremento de fosfatasa alcalina en sangre, incremento de g-glutamiltransferasa, incremento de fosfoquinasa creatina en sangre, incremento de glucosa en sangre, disminución de peso, incremento de peso. No común: disminución de hemoglobina, disminución en la cuenta de neutrófilos, incremento de lactato deshidrogenasa en sangre, disminución de glucosa en sangre, incremento de creatinina en sangre, incremento de urea en sangre. Frecuencia desconocida: incremento de troponina, disminución de potasio en sangre, incremento de bilirrubina no conjugada en sangre, incremento de insulina en sangre, incremento de lipoproteína de muy baja densidad, incremento de la hormona paratiroidea en sangre, disminución de fósforo en sangre, incremento de potasio en sangre, incremento de la presión sanguínea, disminución de la cuenta de células blancas en sangre. Alteraciones de laboratorio: En la tabla 8 se presentan las alteraciones clínicamente importantes o graves de las cifras de laboratorio de hematología o de bioquímica.
Tabla 8. Anormalidades de laboratorio grado ¾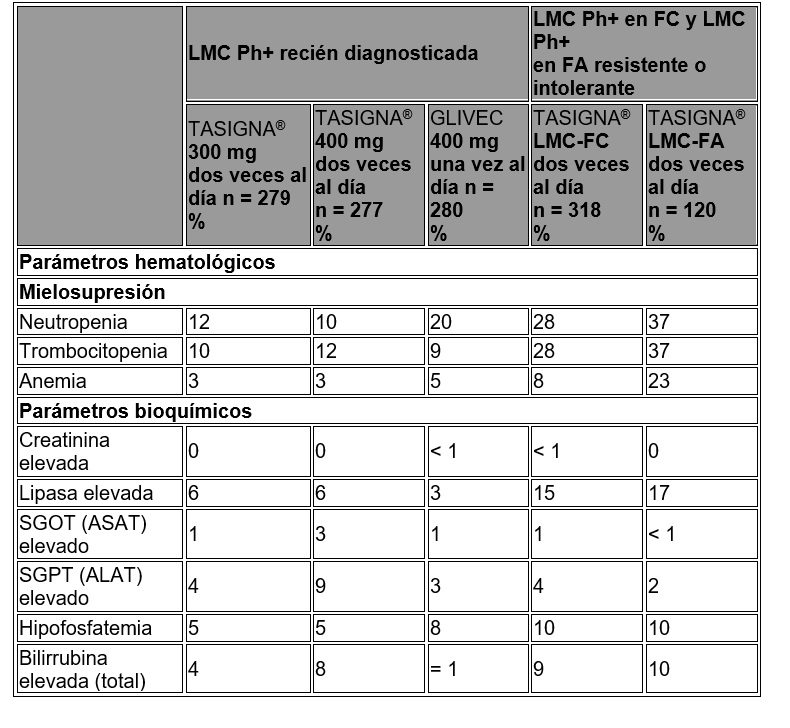
Interacciones medicamentosas y de otro género: Fármacos que pueden aumentar las concentraciones séricas de nilotinib: El nilotinib es principalmente metabolizado en el hígado, y también es un sustrato para la bomba de salida de fármacos-múltiples, como la P-glicoproteína (Pgp). Por lo tanto, la absorción y eliminación subsecuente del nilotinib absorbido sistémicamente puede ser influida por fármacos que afectan el CYP3A4 y/o al Pgp. En un estudio Fase I de nilotinib dado en combinación con imatinib (un sustrato y moderador de Pg-p y CYP3A4), ambos fármacos tuvieron un efecto inhibitorio leve en CYP3A4 y/o PgP. Cuando ambos fármacos fueron administrados concomitantemente, el área bajo la curva de imatinib fue incrementado de 18, 39% y el área bajo la curva de nilotinib fue incrementado de 18 a 40%. La biodisponibilidad de nilotinib en sujetos sanos fue aumentada por tres veces cuando se coadministró con el inhibidor de CYP3A4, el ketoconazol. El tratamiento concomitante con los inhibidores potentes de CYP3A4 deben ser evitados (incluyendo pero no limitando al ketoconazol, itraconazol, voriconazol, ritonavir, claritromicina, y telitromicina) (véase Dosis y vía de administración y Precauciones generales relacionados con la prolongación del intervalo QT). Se debe considerar el uso concomitante de medicamentos alternos que causen poca o mínima inhibición de CYP3A4. Fármacos que pueden disminuir las concentraciones séricas de nilotinib: Los inductores de la actividad de CYP3A4 podrían aumentar el metabolismo del nilotinib y por lo tanto, disminuir sus concentraciones plasmáticas. La administración concomitante de medicamentos que inducen al CYP3A4 (por ejemplo, fenitoína, rifampicina, carbamacepina, fenobarbital y la verruga de St. John) puede disminuir la exposición al nilotinib. En aquellos pacientes en quienes están indicados los inductores de CYP3A4, se debe considerar utilizar agentes alternos con menos potencial de inducción enzimática. En sujetos sanos recibiendo el inductor de CYP3A4 rifampicina a 600 mg por 12 días, la exposición sistémica (ABC) a nilotinib fue disminuida aproximadamente 80%. Fármacos en los cuales se altera la concentración sistémica debido a nilotinib: El nilotinib es un inhibidor competitivo in vitro de CYP3A4, CYP2C8, CYP2C9, CYP2D6 y UGT1A1 in vitro, aumentando potencialmente las concentraciones de fármacos eliminados por estas enzimas. Además, la administración de una dosis única de TASIGNA® con midazolam a sujetos sanos aumentó la exposición de midazolam por 30%. Se debe tener cuidado con la coadministración de TASIGNA® con sustratos de estas enzimas que tienen un índice terapéutico estrecho. Ya que la warfarina es metabolizada por CYP2C9 y CYP3A4, debe de ser evitada si es posible. Otros medicamentos para la anticoagulación deberán ser considerados. Medicamentos antiarrítmicos y otros fármacos que puedan prolongar el QT: El uso concomitante de medicinas antiarrítmicas (incluyendo pero no limitado a amiodarona, disopiramida, procainamida quinidina y sotalol) y otros fármacos que pueden prolongar el intervalo QT (incluyendo pero no limitado a cloroquina, halofantrina, claritromicina, haloperidol, metadona, moxifloxacino, bepridil y pimozide) deben ser evitados (véase Precauciones generales). Otras interacciones que puedan afectar las concentraciones séricas: La absorción de nilotinib aumenta si es tomada con los alimentos, resultando en una concentración sérica más alta (véase Dosis y vía de administración y Precauciones generales). El jugo de toronja y otros alimentos que inhiben CYPA34 deben ser evitados en cualquier momento.
Alteraciones en los resultados de pruebas de laboratorio:
Muy comunes: aumento en la lipasa sérica. Común: aumento en la amilasa sérica, aumento de la alaninotransferasa, aumento en la aspartato aminotranferasa, aumento en la bilirrubina sérica, aumento en la fosfatasa alcalina sérica, y-glutamiltransferasa, aumento de la fosfocinasa creatinina sérica, aumento de la glucosa sérica, disminución de peso, aumento de peso. Poco común: aumento de la lactato deshidrogenasa sérica, disminución de la glucosa sérica, aumento de la creatinina sérica, aumento de la urea sérica. Frecuencia desconocida: aumento de la troponina, disminución del potasio sérico, aumento de la bilirrubina no conjugada sérica. Los valores bioquímicos o hematológicos de rutina clínicamente relevantes o anormalidades severas son presentados en la tabla 9. Tabla 9. Reacciones adversas grado ¾ anormalidades de pruebas de laboratorio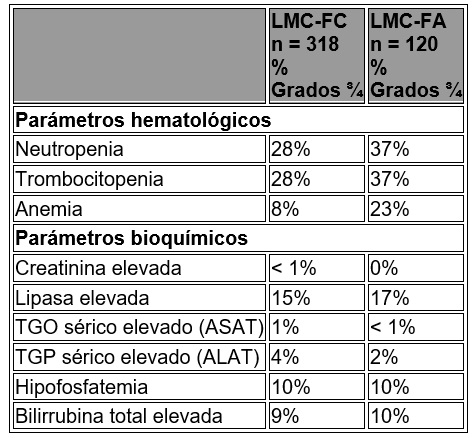
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: El nilotinib ha sido evaluado en estudios de farmacología de seguridad, toxicidad de dosis repetida, genotoxicidad y de toxicidad reproductiva y estudios de fototoxicidad. El nilotinib no tuvo efectos sobre el SNC o las funciones respiratorias. Los estudios de seguridad cardiaca in vitro mostraron una señal preclínica para la prolongación del intervalo QT. No se observaron efectos en las mediciones ECG en perros o monos tratados por hasta 39 semanas o en un estudio de telemetría especial en perros. Los estudios de toxicidad por dosis repetidas en perros por hasta 4 semanas de duración y en monos cinomolgus por hasta 9 meses, revelaron al hígado como órgano blanco primario de toxicidad por nilotinib. Las alteraciones incluyeron un aumento en la actividad de aminotransferasa de alanina y fosfatasa alcalina, y los hallazgos histopatológicos (principalmente hiperplasia/hipertrofia de célula sinusoidal o de célula Kupffer, hiperplasia del conducto biliar y fibrosis periportal). En general, los cambios en la química clínica fueron completamente reversibles después de un periodo de recuperación de cuatro semanas, las alteraciones histológicas solamente mostraron reversibilidad parcial. Las exposiciones a los niveles de dosis más bajos donde los efectos del hígado fueron observados, fueron menores a los de aquéllos de exposición en humanos a una dosis de 800 mg/día. Solamente alteraciones menores del hígado fueron vistos en ratones o ratas tratadas por hasta 26 semanas. Aumentos principalmente reversibles de los niveles de colesterol fueron observados en ratas, perros y monos. Los estudios de genotoxicidad en sistemas bacterianos in vitro y en sistemas mamíferos in vitro e in vivo con o sin activación metabólica, no revelaron evidencia alguna de un potencial mutagénico de nilotinib. El nilotinib no induce teratogenicidad, pero sí mostró embrio y fetotoxicidad a dosis que también mostraron toxicidad materna. Se observó una mayor pérdida de posimplantación en el estudio de fertilidad, tratando a pacientes masculinos y femeninos y en el estudio de embriotoxicidad con el tratamiento de hembras. La letalidad embrionaria y los efectos fetales (principalmente disminución de pesos fetales, variaciones esqueléticas y viscerales) en ratas y una mayor resorción de fetos y variaciones esqueléticas en conejos se encontraron presentes en los estudios de embriotoxicidad. La exposición al nilotinib por hembras en niveles de efecto adverso no observado fue generalmente menor o igual a ese en humanos con dosis de 800 mg/día. En un estudio pre y posnatal, la administración oral de nilotinib a las ratas hembras desde el día 6 de la gestación a los días 21 o 22 posparto resultó en efectos maternos (consumo de alimentos reducido y bajas ganancias de peso corporal) y un periodo de gestación más largo a 60 mg/kg. La dosis materna de 60 mg/kg estuvo asociada en el producto con peso corporal disminuido y cambios en algunos parámetros de desarrollo físico (el día promedio para el desenrollamiento del pabellón auricular, la erupción de dientes y la apertura de ojos fue más temprano). El nivel de efectos adversos no observados en los animales maternos y en sus crías fue una dosis materna de 20 mg/kg. Se mostró que el nilotinib absorbe la luz de los rangos UV-B y UV-A, y a ser distribuido en la piel mostrando un potencial fototóxico in vitro. Sin embargo, no se han observado efectos in vivo. Por lo tanto, el riesgo de que nilotinib cause fotosensibilidad en pacientes es considerado ser muy bajo. No se han realizado estudios de carcinogenicidad con nilotinib.
Precauciones generales: Mielosupresión: El tratamiento con TASIGNA® está a menudo asociado con trombocitopenia, neutropenia y ane