TOPAMAX
MOKSHA8
Denominación genérica: Topiramato
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Topiramato de 25 mg, excipiente cbp. Cada tableta contiene: Topiramato 50 mg, excipiente cbp. Cada tableta contiene: Topiramato 100 mg, excipiente cbp.
Indicaciones terapéuticas: Epilepsia. TOPAMAX® está indicado como: Monoterapia en pacientes con epilepsia de reciente diagnóstico o para la conversión a monoterapia en pacientes con epilepsia. TOPAMAX® está indicado como terapia adjunta en adultos y niños (de 2 años o mayores) con crisis de inicio parcial y crisis generalizadas tónico-clónicas. TOPAMAX® también está indicado en adultos y niños como terapia adjunta en el tratamiento de convulsiones asociadas con el síndrome de Lennox-Gastaut. Migraña.TOPAMAX® está indicado en adultos para el tratamiento profiláctico de la cefalea migrañosa. La utilidad de TOPAMAX® en el tratamiento agudo de cefaleas migrañosas no ha sido estudiada.
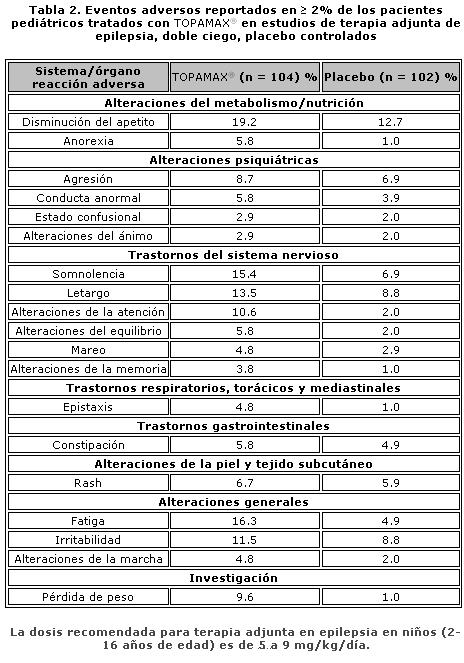
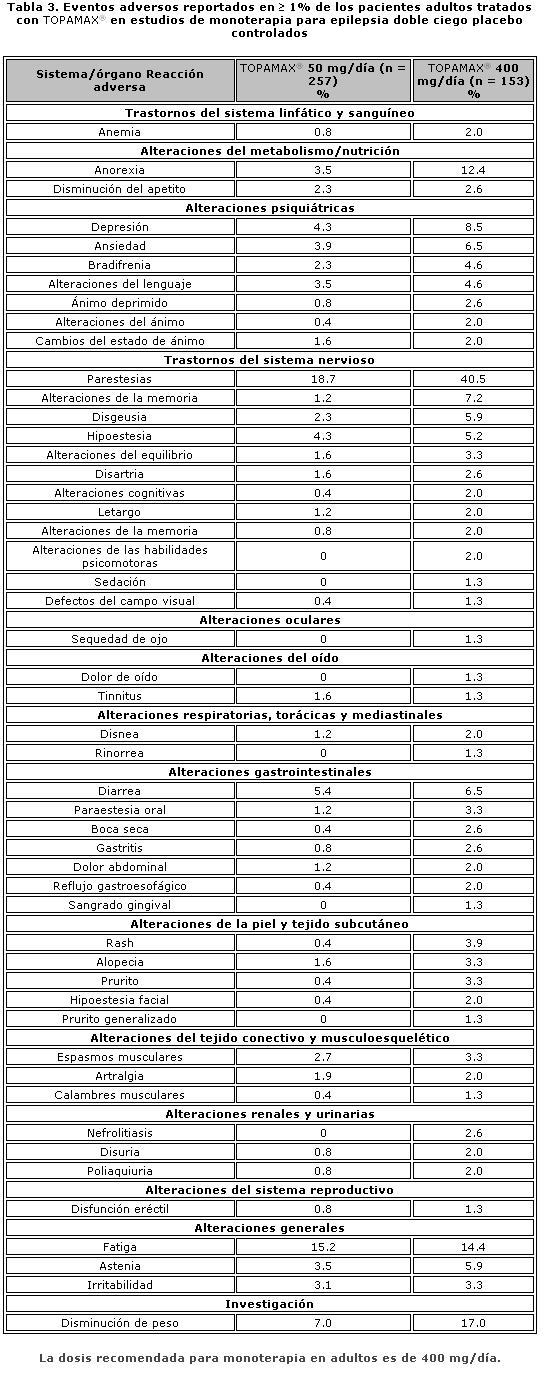
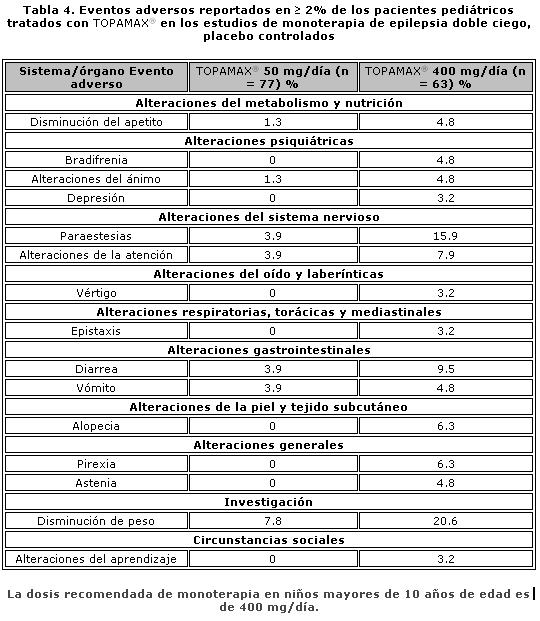
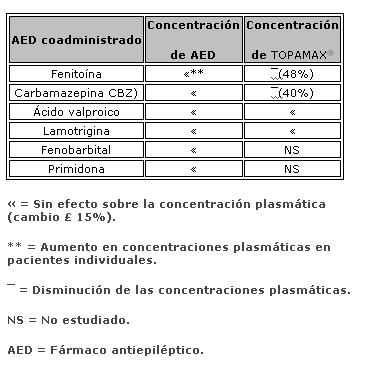
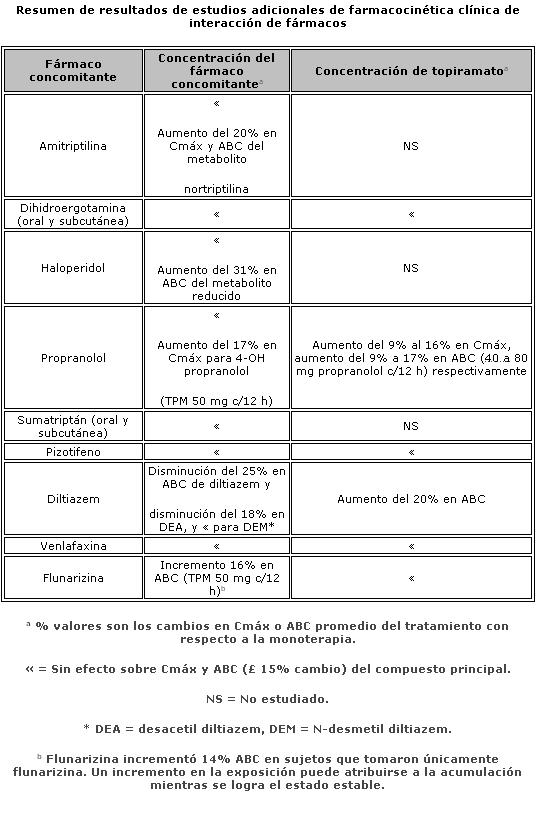
Farmacocinética y farmacodinamia: El nombre químico de topiramato es sulfamato de 2,3:4,5-bis-O-(1-metiletiliden)-b-D-fructopiranosa. La fórmula empírica es C12H21NO8S. El peso molecular es 339.36. Topiramato es un polvo blanco cristalino con sabor amargo. Topiramato es más soluble en soluciones alcalinas que contienen hidróxido de sodio o fosfato de sodio, y que tienen un pH de 9 a 10. Es libremente soluble en acetona, cloroformo, dimetilsulfóxido y etanol. La solubilidad en agua es 9.8 mg/mL. Una solución saturada tiene un pH de 6.3. Propiedades farmacocinéticas. El perfil farmacocinético de topiramato en comparación con otros medicamentos antiepilépticos (MAE) muestra una vida media plasmática larga, farmacocinética lineal, depuración predominantemente renal, ausencia significativa de unión a proteínas y ausencia de metabolitos activos clínicamente relevantes. Topiramato no es inductor potente de enzimas metabolizadoras de fármacos, puede ser administrado independientemente de los alimentos y no es necesario el monitoreo rutinario de concentraciones plasmáticas. En estudios clínicos, no hubo una relación consistente entre las concentraciones plasmáticas y la eficacia o eventos adversos. Absorción. Topiramato se absorbe bien y de manera rápida. Después de la administración oral de 100 mg de topiramato a sujetos sanos, se alcanzó una concentración plasmática pico promedio (Cmax) de 1.5 mcg/mL dentro de un periodo de tiempo de 2 a 3 horas (Tmáx). En base a la recuperación de la radioactividad en la orina, el grado de absorción promedio de una dosis oral de 100 mg de 14C-topiramato fue al menos del 81%. No existió efecto clínicamente significativo de la comida sobre la biodisponibilidad de topiramato. Distribución. Generalmente de 13% a 17% de topiramato está unido a proteínas plasmáticas. Se ha observado una baja capacidad en los sitios de unión de los eritrocitos para topiramato, los cuales se saturan por encima de concentraciones plasmáticas de 4 mcg/mL. El volumen de distribución varía inversamente con la dosis. El volumen aparente promedio de distribución fue 0.8 a 0.55 L/kg para dosis únicas varía de 100 a 1,200 mg. Se detectó un efecto del género sobre el volumen de distribución, siendo los valores para mujeres cerca de 50% de aquellos de los hombres. Esto se atribuye al mayor porcentaje de grasa corporal en mujeres y no tiene consecuencias clínicas. Metabolismo. Topiramato no se metaboliza en forma extensa (~20%) en voluntarios sanos. Se metaboliza hasta en un 50% en pacientes que reciben terapia antiepiléptica concomitante con inductores conocidos de enzimas metabolizadoras de fármacos. Se han aislado seis metabolitos producto de la hidroxilación, hidrólisis y glucuronidación, caracterizados e identificados a partir de plasma, orina y heces en seres humanos. Cada metabolito representa menos del 3% de la radioactividad total excretada después de la administración de 14C-topiramato. Dos metabolitos, que conservan gran parte de la estructura de topiramato, fueron analizados y se encontró que tenían poca o ninguna actividad anticonvulsiva. Eliminación. En humanos, la principal ruta de eliminación de topiramato sin cambios y sus metabolitos es por medio del riñón (por lo menos el 81% de la dosis). Aproximadamente 66% de una dosis de 14C-topiramato fue excretada sin cambios en la orina en un periodo de cuatro días. Después de una dosis de 50 mg y 100 mg de topiramato dos veces al día, la depuración renal media fue de aproximadamente 18 mL/min y 17 mL/min respectivamente. Existe evidencia de reabsorción tubular renal de topiramato. Esto está soportado por estudios de ratas en donde topiramato fue coadministrado con probenecid, y se observó un aumento significativo en la depuración renal de topiramato. En general, la depuración plasmática es aproximadamente de 20 a 30 mL/min en humanos después de la administración oral. Topiramato presenta baja variabilidad inter-sujeto en las concentraciones plasmáticas y, por lo tanto, tiene una farmacocinética predecible. La farmacocinética de topiramato es lineal con la depuración plasmática permaneciendo constante y el área bajo la curva de la concentración plasmática aumentando de manera proporcional a la dosis en un rango de dosis de 100 a 400 mg de dosis individuales en sujetos sanos. Los pacientes con función renal normal pueden tardar de 4 a 8 días en alcanzar concentraciones plasmáticas de estado estacionario. La Cmax media, después de la administración de dosis orales múltiples de 100 mg dos veces al día, en sujetos sanos fue de 6.76 mcg/mL. Después de la administración de dosis múltiples de 50 mg y 100 mg de topiramato dos veces al día, la vida media de eliminación plasmática fue aproximadamente 21 horas. Uso con otros MAE. La administración de dosis múltiples de topiramato, 100 a 400 mg dos veces al día, de manera concomitante con fenitoína o carbamazepina muestra incrementos en las concentraciones plasmáticas de topiramato proporcionales a la dosis. Poblaciones especiales. Pediátrica (hasta 12 años de edad). La farmacocinética de topiramato en niños, al igual que en los adultos que reciben terapia coadyuvante, es lineal, con la depuración independiente de la dosis y de las concentraciones plasmáticas en estado estacionario aumentando en proporción a la dosis. Los niños, sin embargo, tienen una depuración más alta y una vida media de eliminación más corta. En consecuencia, las concentraciones plasmáticas de topiramato para la misma dosis mg/kg pueden ser menores en niños en comparación con adultos. Al igual que en adultos, los MAE inductores de enzimas hepáticas disminuyen las concentraciones plasmáticas en estado estacionario. Adultos mayores. La depuración plasmática de topiramato no cambia en sujetos adultos mayores, en ausencia de un padecimiento renal subyacente. Insuficiencia renal. La depuración plasmática y renal de topiramato disminuye en pacientes con daño moderado y severo en la función renal (ClCR < 70 mL/min). Como resultado, se esperan concentraciones plasmáticas de topiramato en estado estacionario más altas para una dosis dada en pacientes con insuficiencia renal en comparación con aquellos con una función renal normal. Además, los pacientes con insuficiencia renal requerirán un tiempo más prolongado para alcanzar el estado estacionario con cada dosis. En pacientes con insuficiencia renal moderada y severa, se recomienda la mitad de la dosis habitual inicial y de mantenimiento (ver Dosis y vía de administración - Poblaciones especiales, Insuficiencia renal). Topiramato es removido efectivamente del plasma por hemodiálisis. Un período prolongado de hemodiálisis puede causar que la concentración de topiramato disminuya por debajo de los niveles que se requieren para mantener el efecto anticonvulsivo. A fin de evitar rápidas disminuciones en la concentración plasmática de topiramato durante la hemodiálisis, se puede requerir una dosis complementaria de topiramato. El ajuste real debe tener en cuenta 1) la duración del período de diálisis, 2) el índice de eliminación del sistema de diálisis que se está utilizando, y 3) la depuración renal efectiva de topiramato en el paciente a ser dializado. Insuficiencia hepática. La depuración plasmática de topiramato disminuye en una media del 26% en pacientes con insuficiencia hepática moderada a severa. Por lo tanto, topiramato debe administrarse con precaución en pacientes con insuficiencia hepática. Propiedades farmacodinámicas. Grupo farmacoterapéutico: otros antiepilépticos, código ATC: N03AX11. Topiramato está clasificado como un monosacárido - sulfamato sustituido. Se desconoce el mecanismo exacto a través del cual topiramato ejerce su efecto anticonvulsivo y para profilaxis de la migraña. Mediante estudios electrofisiológicos y bioquímicos en neuronas cultivadas se han identificado tres propiedades que potencialmente pudieran contribuir a la eficacia antiepiléptica de topiramato. Topiramato bloqueó los potenciales de acción generados de forma repetitiva a través de una despolarización neuronal sostenida de forma dependiente del tiempo, lo cual sugiere una acción de bloqueo de los canales de sodio estado dependiente. Topiramato incrementa la frecuencia a la cual el ácido c-aminobutírico (GABA) activa a los receptores GABAA, y mejoró la habilidad del GABA para inducir el flujo de iones de cloro a la neurona, sugiriendo que topiramato potencia la actividad de este neurotransmisor inhibitorio. Este efecto no fue bloqueado por flumazenil, un antagonista de las benzodiacepinas, tampoco aumentó la duración del tiempo de apertura de canal, diferenciando topiramato de los barbitúricos que modulan los receptores GABAA. Debido a que el perfil antiepiléptico de topiramato difiere notoriamente del de las benzodiacepinas, éste pudiera modular un subtipo de receptor GABAA no sensible a benzodiacepinas. El topiramato antagonizó la habilidad del kainato para activar al receptor kainato/AMPA (ácido a-amino-3-hidroxi-5-metilisoxazol-4 propiónico), que es un subtipo de receptor del aminoácido excitatorio (glutamato), pero no tiene un efecto aparente sobre la actividad del N-metil-D-aspartato (NMDA) en el subtipo de receptor de NMDA. Estos efectos de topiramato fueron dependientes de la concentración en un intervalo de 1 mcM a 200 mcM, con actividad mínima observada en 1 mcM a 10 mcM. Además, topiramato inhibe a algunas de las isoenzimas de la anhidrasa carbónica. Este efecto farmacológico es mucho más débil que el de acetazolamida, un inhibidor de la anhidrasa carbónica conocido y no se piensa que sea un componente importante de la actividad antiepiléptica de topiramato. En estudios animales, topiramato tiene una actividad anticonvulsiva en las pruebas de crisis convulsiva por "electroshock" en ratas y ratones (MES, por sus siglas en inglés "mouse maximal electroshock seizure) y es efectivo en los modelos de epilepsia en roedores, lo cual incluye a las crisis tónicas y de ausencia presentes en las ratas con crisis epilépticas espontáneas (SER, por sus siglas en inglés "spontaneous epileptic rat") y en las crisis tónico clónicas inducidas en ratas a través de estimulación eléctrica repetida (kindling) en la amígdala o por isquemia global. Topiramato es sólo débilmente efectivo en el bloqueo de las convulsiones clónicas inducidas por el antagonista del receptor GABAA, pentilentetrazol. En los estudios en ratones en los cuales se administró topiramato y carbamacepina o fenobarbital de manera concomitante, se observó una actividad anticonvulsiva sinérgica, mientras que con la combinación con fenitoína se obtuvo una actividad anticonvulsiva aditiva. En los estudios controlados de terapia adjunta, no se ha demostrado una correlación entre la concentración plasmática mínima de topiramato y su eficacia clínica. No se ha demostrado evidencia de tolerancia en el hombre. Estudios clínicos sobre epilepsia Los resultados de estudios clínicos controlados establecieron la eficacia de TOPAMAX® tabletas como monoterapia para adultos y niños (6 años de edad y mayores) con epilepsia, como terapia coadyuvante en adultos y pacientes pediátricos de 2 a 16 años de edad con crisis parciales de inicio o convulsiones tónico clónicas primariamente generalizadas, y en pacientes de 2 años de edad y mayores con convulsiones asociadas a síndrome de Lennox-Gastaut.Monoterapia La efectividad de topiramato como monoterapia en adultos y niños de 6 años de edad y mayores con reciente diagnóstico de epilepsia fue establecida en 4 estudios aleatorizados, paralelos, doble ciego. El estudio EPMN-106 fue conducido en 487 pacientes (6 a 83 años de edad) quienes tuvieron diagnóstico reciente de epilepsia (parcial de inicio o generalizada) o un diagnóstico de epilepsia recurrente mientras no consumiera MAE. Los pacientes fueron aleatorizados para recibir topiramato 50 mg/día o topiramato 400 mg/día. Los pacientes permanecieron en la fase doble ciego hasta que experimentaban una primera crisis parcial o convulsión tónico-clónica generalizada, hasta el término de la fase doble ciego 6 meses después de la aleatorización del último sujeto, o hasta el abandono por razones específicas del protocolo. La valoración de eficacia primaria fue basada en la comparación entre los grupos con dosis de topiramato con respecto al tiempo de aparición de la primera crisis parcial o convulsión tónico-clónica generalizada durante la fase doble-ciego. La comparación de las curvas de supervivencia de Kaplan-Meier del tiempo de aparición de la primera convulsión favoreció a topiramato 400 mg/día sobre topiramato 50 mg/día (p=0.0002, prueba log rank). La separación entre grupos en favor del grupo con dosis más altas ocurrió tempranamente en la fase de ajuste de la dosis y fue estadísticamente significativa tan pronto como a las 2 semanas post-aleatorización (p=0.046), cuando, siguiendo el esquema semanal de ajuste de dosis, los sujetos en el grupo con la dosis mayor alcanzaron una dosis máxima de topiramato de 100 mg/día. El grupo con dosis mayores también fue superior comparado con el grupo de dosis más bajas con respecto a la proporción de sujetos que permanecieron sin convulsiones, basados en estimaciones de Kaplan Meier, por un mínimo de 6 meses de terapia (82.9% contra 71.4%; p= 0.005), y por un mínimo de 1 año de terapia (75.7% contra 58.8%; p=0.001). La proporción de la tasa de riesgo en el tiempo para la aparición de la primera convulsión fue de 0.516 (Intervalo de confianza 95%, 0.364 a 0.733). Los efectos del tratamiento con respecto al tiempo para la aparición de la primera convulsión fueron consistentes a lo largo de varios subgrupos de sujetos definidos por edad, sexo, región geográfica, peso corporal basal, tipo de convulsión basal, tiempo transcurrido desde el diagnóstico y uso de fármaco antiepiléptico basal. En el estudio YI, un estudio monocéntrico, en pacientes con edades de los 15 a los 63 años, con convulsiones parciales de inicio refractarias (n=48) fueron cambiados de su tratamiento existente a TOPAMAX® 100 mg/día o 1000 mg/día como monoterapia. El grupo con dosis altas fue estadísticamente superior al grupo de dosis bajas por variables de eficacia. 54% de los pacientes con dosis altas cumplieron la monoterapia comparados con 17% en el grupo de dosis bajas con la diferencia entre las dosis siendo estadísticamente significativa (p=0.005). La media de tiempo para la salida fue significativamente superior en el grupo con dosis altas (p=0.002). Las evaluaciones del investigador y globales del sujeto para la respuesta clínica favorecieron estadísticamente al grupo con dosis altas (≤0.002). En el estudio EPMN-104, los pacientes pediátricos y adultos (6-85 años de edad) con diagnóstico reciente de epilepsia (n=252) fueron aleatorizados en los grupos de dosis bajas (25 a 50 mg/día) o de dosis altas (200 o 500 mg/día) basados en su peso corporal. En total, 54% de los pacientes con dosis altas y 39% de los pacientes con dosis bajas fueron reportados libres de convulsiones durante la fase de doble ciego (p=0.022). El grupo con dosis altas fue también superior al de dosis bajas con respecto a la distribución en la frecuencia de las convulsiones (p=0.008) y en la diferencia en cuanto al tiempo transcurrido hasta la aparición de la primera convulsión a lo largo de tres estratos de concentraciones plasmáticas de topiramato (p=0.015). En el estudio EPMN-105, pacientes con edades de los 6-84 años con diagnóstico reciente de epilepsia (n=613) fueron aleatorizados para recibir tanto 100 o 200 mg/día de TOPAMAX® o tratamiento antiepiléptico estándar (carbamazepina o valproato). TOPAMAX® fue por lo menos tan eficaz como carbamazepina o valproato en la disminución de convulsiones en estos pacientes; los intervalos de confianza del 95% para la diferencia entre los dos grupos de tratamiento fueron estrechos e incluyeron el cero, indicando que no existía diferencia estadísticamente significativa entre los grupos. Los dos grupos de tratamiento fueron incluso comparables con respecto a todos los criterios de valoración de eficacia y utilidad clínica incluyendo tiempo para la salida, proporción de sujetos libres de convulsiones y tiempo transcurrido a la primera convulsión. Los pacientes (n=207; 32 con edades ≤16 años) quienes completaron la fase doble ciego del estudio YI y EPMN-104 fueron reclutados en estudios de extensión a largo plazo con la mayoría de los pacientes recibiendo TOPAMAX® durante 2 a 5 años. En esos estudios, se demostró eficacia sostenida con la administración a largo plazo de TOPAMAX® como monoterapia. No existió algún cambio significativo en la dosificación durante el período de extensión y no hubo indicación de la efectividad de TOPAMAX® como monoterapia disminuida con exposición continua. Terapia adyuvante. Estudios controlados en pacientes con convulsiones parciales de inicio. Adultos con convulsiones parciales de inicio. La efectividad de topiramato como tratamiento adyuvante para adultos con convulsiones parciales de inicio fue establecida en seis estudios multicéntricos, aleatorizados, doble ciego, controlados con placebo, dos estudios comparando varias dosis de topiramato y placebo, y cuatro comparando dosis únicas con placebo en pacientes con historial de crisis parciales de inicio, con o sin convulsiones secundariamente generalizadas.A los pacientes en estos estudios se les permitió un máximo de dos MAE adicionales a TOPAMAX® tabletas o placebo. En cada estudio, los pacientes fueron estabilizados a dosis óptimas de sus MAE concomitantes durante la fase basal con duración de 4 a 12 semanas. Los pacientes que experimentaban un número mínimo preespecificado de convulsiones parciales de inicio, con o sin generalización secundaria, durante la fase basal (12 convulsiones para la base de 12 semanas, 8 para la base de 8 semanas, o 3 para la base 4 semanas) fueron asignados de manera aleatoria a placebo o a dosis específica de TOPAMAX® tabletas en adición a sus otros MAE. Después de la aleatorización, los pacientes iniciaron la fase doble ciego del tratamiento. En cinco de los seis estudios, los pacientes recibieron el medicamento activo comenzando a 100 mg por día; la dosis se incrementó posteriormente en 100 mg o 200 mg/día semanalmente o cada 2 semanas hasta que la dosis asignada se alcanzaba, a menos que la intolerancia previniera incrementos. En el sexto estudio (119), las dosis iniciales de 25 o 50 mg/día de topiramato continuaron con sus respectivos incrementos semanales de 25 o 50 mg/día hasta alcanzar las dosis meta de 200 mg/día. Después del ajuste de la dosis, los pacientes entraron en un período de estabilización de 4, 8 o 12 semanas. Los números de pacientes aleatorizados a cada dosis, y la dosis media y mediana real en el período de estabilización se muestran en las tablas 2 y 3. Pacientes pediátricos de 2 a 16 años de edad con convulsiones parciales de inicio. La efectividad de topiramato como tratamiento coadyuvante para pacientes pediátricos en edades de los 2 a 16 años de edad con convulsiones parciales de inicio fue establecida en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, que comparaba topiramato y placebo en pacientes con historia de crisis parciales de inicio, con o sin convulsiones secundariamente generalizadas. Los pacientes en este estudio tenían permitido recibir un máximo de dos MAE adicionales a TOPAMAX® tabletas o placebo. En este estudio, los pacientes fueron estabilizados a dosis óptimas de sus MAE concomitantes durante una fase basal de 8 semanas. Los pacientes que experimentaron por lo menos seis crisis parciales de inicio, con o sin convulsiones secundariamente generalizadas, durante la fase basal fueron asignados aleatoriamente a placebo o TOPAMAX® tabletas en adición a sus otros MAE. Posterior a la aleatorización, los pacientes iniciaron la fase doble ciego del tratamiento. Los pacientes recibieron el medicamento activo iniciando con 25 o 50 mg por día; la dosis fue aumentada en incrementos de 25 mg a 150 mg/día cada 2 semanas hasta alcanzar la dosis asignada de 125, 175, 225 o 400 mg/día basada en el peso de los pacientes hasta alcanzar a una dosis de 6 mg/kg/día, a menos que la intolerancia previniera los incrementos. Después del ajuste, los pacientes entraron en un período de estabilización de 8 semanas. Estudios controlados en pacientes con convulsiones tónico-clónicas primariamente generalizadas. La efectividad de topiramato como tratamiento adyuvante para convulsiones tónico-clónicas primariamente generalizadas en pacientes de 2 años de edad y mayores fue establecida en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, que comparaba dosis únicas de topiramato y placebo. Los pacientes en este estudio podían recibir un máximo de dos MAE adicionales a TOPAMAX® o placebo. Los pacientes fueron estabilizados a dosis óptimas de sus MAE concomitantes durante una fase basal en 8 semanas. Los pacientes que experimentaron por lo menos tres crisis convulsivas tónico-clónicas primariamente generalizadas durante la fase basal fueron asignados de manera aleatoria a placebo o TOPAMAX® en adición a sus otros fármacos antiepilépticos. Posterior a la aleatorización, los pacientes iniciaron una fase doble ciego del tratamiento. Los pacientes recibieron el medicamento activo iniciando con 50 mg/día para cuatro semanas; la dosis se incrementó posteriormente en incrementos de 50 a 150 mg/día cada 2 semanas hasta la dosis asignada de 175, 225 o 400 mg/día basado en el peso corporal del paciente hasta aproximarse a dosis de 6 mg/kg por día, a menos que la intolerancia previniera el incremento. Después del ajuste, los pacientes entraron en un período de estabilización de 12 semanas. Estudios controlados en pacientes con Síndrome de Lennox-Gastaut. La efectividad de topiramato como tratamiento adyuvante para las convulsiones asociadas con el síndrome de Lennox-Gastaut fue establecido en un estudio multicéntrico, aleatorizado, doble ciego, controlado por placebo comparando una sola dosis de topiramato con placebo en pacientes de 2 años de edad o mayores. Los pacientes en este estudio podían recibir un máximo de dos MAE adicionales a TOPAMAX® o placebo. Los pacientes que experimentaban por lo menos 60 convulsiones mensuales antes de entrar al estudio fueron estabilizados en dosis óptimas de sus MAE concomitantes durante una fase basal de cuatro semanas. Posterior al estado basal, los pacientes fueron asignados aleatoriamente a placebo o TOPAMAX® en adición a sus otros MAE. El medicamento activo fue ajustado iniciando con 1 mg /kg/día durante una semana; la dosis fue aumentada a 3 mg/kg/día durante una semana y luego a 6 mg/kg/día. Después del ajuste de dosis, los pacientes entraron en un período de estabilización de 8 semanas. Las mediciones primarias de efectividad fueron el porcentaje de reducción en crisis que provocan caídas y una escala parental global de la intensidad de las convulsiones. En todos los estudios complementarios se midió la reducción en la tasa de convulsiones desde la basal durante toda la fase de doble ciego. La mediana del porcentaje de reducciones en las tasas de convulsión y las tasas de respuesta (fracción de los pacientes con por lo menos 50% de reducción) por grupo de tratamiento para cada estudio se muestran a continuación en la Tabla 1. Como se describió previamente, la mejoría global en la severidad de las convulsiones también fue valorada en el estudio de Lennox-Gastaut.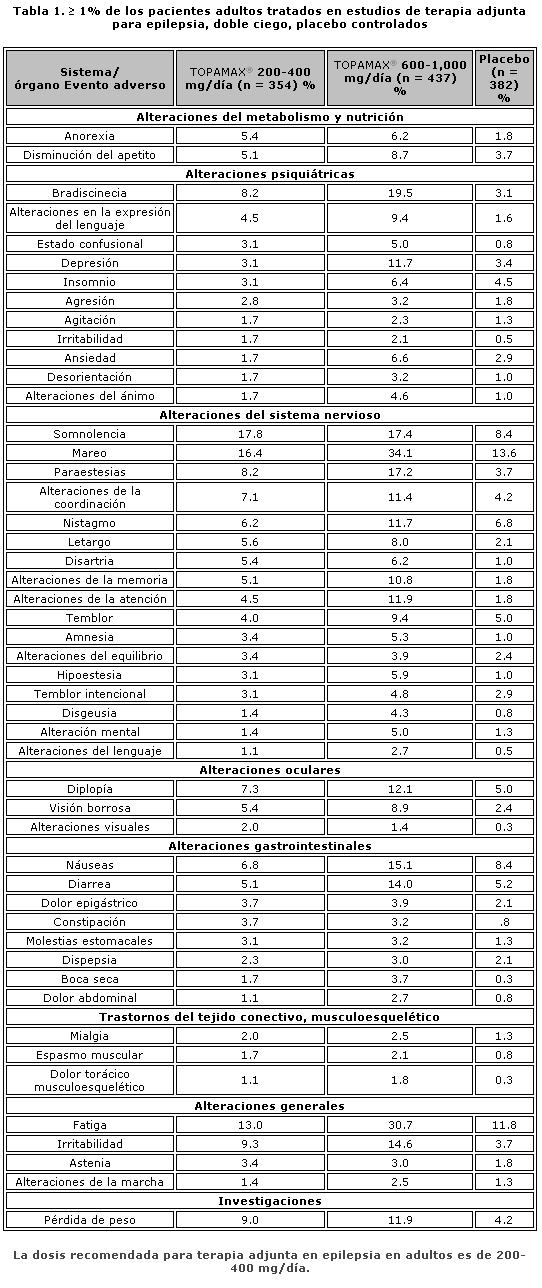
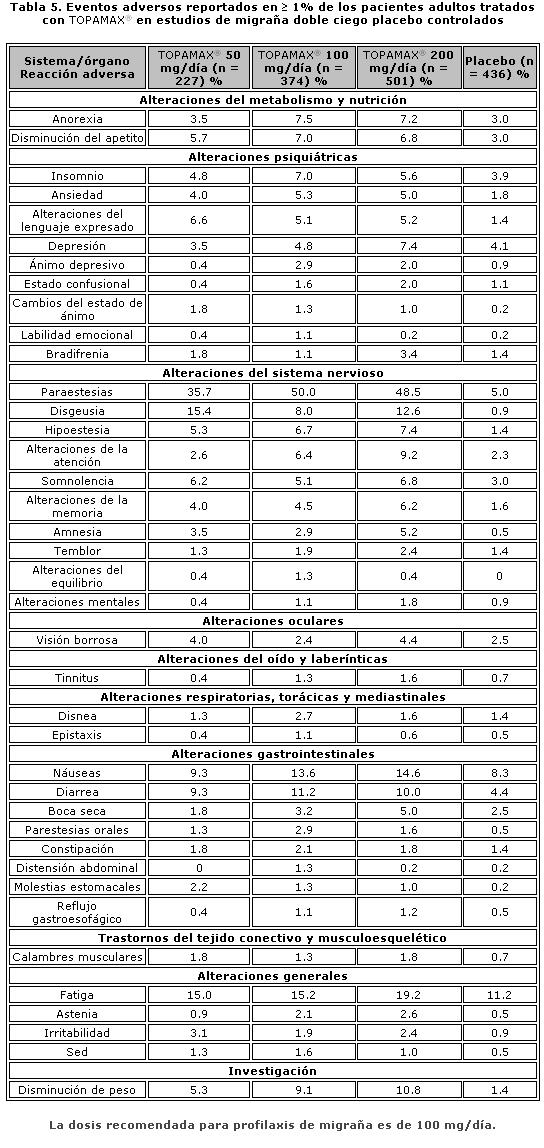
Los análisis de los subconjuntos de la eficacia antiepiléptica de TOPAMAX® tabletas en estos estudios no mostró diferencias en función del género, raza, edad, tasa basal de convulsiones, o MAE concomitantes. Estudios clínicos de migraña. El programa de desarrollo clínico para evaluar la eficacia de TOPAMAX® en la profilaxis de migraña incluyó dos estudios pivote multicéntricos, aleatorizados, doble ciego, controlados con placebo, paralelos, conducidos en Norte América (MIGR-001 y MIGR-002). El objetivo primario de la eficacia fue la reducción de la frecuencia de la cefalea migrañosa, medida por el cambio en la tasa de 4 semanas de migraña de la fase basal a la fase de tratamiento doble ciego en cada grupo de tratamiento con TOPAMAX® comparado con el placebo en la población que se intentó de tratar (ITT por sus siglas en inglés). Los resultados agrupados de los dos estudios pivote evaluando las dosis de TOPAMAX® de 50 (N=233), 100 (N=244) y 200 mg/día (N=228) encontraron la reducción del porcentaje mediano en la tasa del período mensual promedio de migraña de 35%, 51% y 49%, respectivamente, comparada con el 21% para el grupo placebo (N=229). Los 100 y 200 mg/día de TOPAMAX® fueron estadísticamente mejores que el placebo. Notablemente, 27% de los pacientes que recibieron TOPAMAX® 100 mg/día alcanzaron por lo menos el 75% de reducción en la frecuencia de migraña, mientras que el 52% alcanzaron por lo menos un 50% de reducción. Un estudio de soporte adicional, el MIGR-003, demostró que TOPAMAX® 100 mg/día fue comparable en términos de eficacia con propanolol 160 mg/día. No existió diferencia estadísticamente significativa entre los dos grupos en el objetivo primario de eficacia.
Contraindicaciones: Hipersensibilidad a cualquiera de los componentes de este producto.
Precauciones generales: Suspensión de TOPAMAX®. En pacientes con o sin historia de convulsiones o epilepsia, los MAE, incluyendo TOPAMAX®, deben de retirarse gradualmente para minimizar el potencial de convulsiones o de que se incremente la frecuencia de convulsiones. En estudios clínicos, las dosis diarias se redujeron en intervalos semanales por 50 a 100 mg en adultos con epilepsia y por 25 a 50 mg en adultos que recibieron TOPAMAX® en dosis de hasta 100 mg/día para profilaxis de migraña. En estudios clínicos en niños, TOPAMAX® se retiró gradualmente en un periodo de 2 a 8 semanas. En situaciones en las que se requiere el rápido retiro de TOPAMAX® por indicación médica, se recomienda un monitoreo apropiado. Insuficiencia renal. La principal vía de eliminación de topiramato sin cambios y sus metabolitos es el riñón. La eliminación renal es dependiente de la función renal y es independiente de la edad. Los pacientes con insuficiencia renal moderada o severa pueden requerir de 10 a 15 días para alcanzar concentraciones plasmáticas en estado estacionario, comparado con 4 a 8 días en pacientes con función renal normal. Al igual que todos los pacientes, el programa de titulación se debe guiar por los resultados clínicos (es decir, control de convulsiones, evitando efectos adversos) con el conocimiento de que los pacientes con insuficiencia renal conocida pueden requerir un tiempo mayor para alcanzar el estado estacionario en cada dosis (ver Dosis y vía de administración - Poblaciones especiales, Insuficiencia renal y Farmacocinética y farmacodinamia - Propiedades farmacocinéticas - Poblaciones especiales, Insuficiencia renal). Hidratación. Se ha reportado oligohidrosis (reducción de la sudoración) y anhidrosis asociados con el uso de topiramato. La reducción de la sudoración e hipertermia (aumento en la temperatura corporal) pueden ocurrir especialmente en niños pequeños expuestos a altas temperaturas ambientales (ver Reacciones secundarias y adversas). Es muy importante la hidratación adecuada mientras se toma topiramato. La hidratación puede reducir el riesgo de nefrolitiasis (ver Precauciones generales - Nefrolitiasis). La hidratación adecuada antes y durante las actividades como ejercicio o la exposición a temperaturas altas puede reducir el riesgo de eventos adversos relacionados con el calor (ver Reacciones secundarias y adversas). Alteraciones del estado de ánimo/depresión. Se ha observado un aumento en la incidencia de alteraciones del estado de ánimo y depresión durante el tratamiento con topiramato. Suicidio/ideación suicida. Los MAE, incluyendo TOPAMAX®, incrementan el riesgo de pensamientos o conductas suicidas en los pacientes que toman estos medicamentos por cualquier indicación. Un metaanálisis que incluyó estudios de MAE, aleatorizados, controlados con placebo, mostró un incremento en el riesgo de ideación y conducta suicida (0.43% para los MAE en comparación con 0.24% para placebo). El mecanismo de este riesgo se desconoce. En los estudios clínicos doble ciego, los eventos relacionados con suicidio (ideación suicida, intentos de suicidio y suicidio) ocurrieron con una frecuencia de 0.5% en los pacientes tratados con topiramato (46 de 8,652 pacientes tratados) comparado con el 0.2% de los pacientes tratados con placebo (8 de 4,045 pacientes tratados). Se reportó un caso de suicidio en un paciente que recibía topiramato dentro de un estudio doble ciego sobre trastorno bipolar. Por esta razón los pacientes deben de ser monitoreados para identificar signos de ideación y conducta suicida y considerar el tratamiento apropiado. Debe recomendarse a los pacientes (y cuando sea conveniente, a los cuidadores de los pacientes) que busquen evaluación médica inmediata si aparecen signos de comportamiento o ideación suicida. Nefrolitiasis. Algunos pacientes, especialmente aquellos con predisposición a la nefrolitiasis, pueden estar en mayor riesgo de formación de cálculos renales y signos y síntomas asociados tales como cólicos renales, dolor renal o dolor en el costado. Los factores de riesgo para nefrolitiasis incluyen formación de cálculos previos, historia familiar de nefrolitiasis e hipercalciuria ver (Precauciones generales - Acidosis metabólica). Ninguno de estos factores de riesgo puede realmente predecir la formación de cálculos durante el tratamiento con topiramato. Además, en los pacientes que están tomando otros medicamentos asociados con nefro-litiasis se puede incrementar el riesgo (ver Interacciones medicamentosas y de otro género - Otras formas de interacción - Agentes que predisponen a nefrolitiasis). Insuficiencia hepática En pacientes con insuficiencia hepática, topiramato se debe administrar con precaución ya que la depuración de topiramato puede disminuir (ver Dosis y vía de administración - Poblaciones especiales, Insuficiencia hepática y Farmacocinética y farmacodinamia - Propiedades farmacocinéticas - Poblaciones especiales, Insuficiencia hepática). Miopía aguda y glaucoma secundario de ángulo cerrado. Se ha reportado un síndrome que consiste en miopía aguda asociada con glaucoma secundario de ángulo cerrado en pacientes que recibieron TOPAMAX®. Los síntomas incluyen un inicio agudo de la reducción de la agudeza visual y/o dolor ocular. Los hallazgos oftalmológicos pueden incluir miopía, estrechamiento de la cámara anterior, hiperemia ocular (enrojecimiento) y aumento en la presión intraocular. Puede o no haber midriasis. Este síndrome puede estar asociado con efusión supraciliar resultando en desplazamiento anterior de la lente e iris con glaucoma secundario de ángulo cerrado. Los síntomas ocurren típicamente dentro de 1 mes del inicio de la terapia con TOPAMAX®. En contraste con el glaucoma primario de ángulo estrecho, que es raro en personas menores de 40 años de edad, el glaucoma secundario de ángulo cerrado asociado con topiramato se ha reportado en pacientes pediátricos y en adultos. El tratamiento incluye la descontinuación de TOPAMAX® tan rápido como sea posible a juicio del médico tratante, y medidas apropiadas para reducir la presión intraocular. Estas medidas generalmente resultan en una disminución de la presión intraocular. La presión intraocular elevada de cualquier etiología, si no es tratada, puede producir secuelas serias incluyendo pérdida permanente de la visión. Defectos del campo visual. Se han reportado defectos del campo visual en pacientes que recibieron topiramato, independiente de la presión intraocular elevada. En los estudios clínicos, la mayoría de estos eventos fueron reversibles después de suspender topiramato. Si se presentan problemas visuales en cualquier momento durante el tratamiento con topiramato, debe considerarse suspender el fármaco. Acidosis metabólica. La acidosis metabólica, hiperclorémica, de brecha no aniónica (es decir, bicarbonato sérico disminuido por debajo del rango de referencia normal en la ausencia de alcalosis respiratoria) está asociada al tratamiento con topiramato. Esta disminución en el bicarbonato sérico es debido al efecto inhibitorio de topiramato sobre la anhidrasa carbónica renal. Generalmente, la disminución en el bicarbonato ocurre de manera temprana en el tratamiento, aunque puede ocurrir en cualquier momento durante el tratamiento. Estas disminuciones son normalmente leves a moderadas (disminución promedio de 4 mmol/L a dosis de 100 mg/día o mayor en adultos y aproximadamente 6 mg/kg/día en pacientes pediátricos). Raramente, los pacientes han experimentado disminuciones a valores por debajo de 10 mmol/L. Las condiciones o terapias que predisponen a la acidosis (tales como enfermedad renal, trastornos respiratorios severos, estado epiléptico, diarrea, cirugía, dieta cetogénica, o ciertos fármacos) pueden ser aditivos a los efectos de topiramato sobre la disminución del bicarbonato. La acidosis metabólica crónica no tratada puede incrementar el riesgo de nefrolitiasis o nefrocalcinosis (ver Precauciones generales - Nefrolitiasis). La acidosis metabólica crónica en pacientes pediátricos puede reducir las tasas de crecimiento. El efecto de topiramato sobre el crecimiento y las secuelas relacionadas con los huesos no han sido sistemáticamente investigadas en poblaciones pediátricas o de adultos. Dependiendo de las condiciones subyacentes, con la terapia con topiramato se recomienda la evaluación apropiada, incluyendo los niveles séricos de bicarbonato. Si la acidosis metabólica se desarrolla y persiste, se deberá considerar reducir la dosis o descontinuar topiramato (utilizando disminución gradual de la dosis). Hiperamonemia y encefalopatía. Se ha reportado hiperamonemia con o sin encefalopatía con el tratamiento con topiramato (ver Reacciones secundarias y adversas). El riesgo de hiperamonemia con topiramato parece estar relacionado con la dosis. Se ha reportado hiperamonemia con más frecuencia cuando topiramato se utiliza concomitantemente con ácido valproico (ver Interacciones medicamentosas y de otro género). Los síntomas clínicos de la encefalopatía hiperamonémica con frecuencia incluyen alteraciones agudas en el nivel de conciencia y/o la función cognitiva con letargia. En la mayoría de los casos, la encefalopatía hiperamonémica disminuyó con la suspensión del tratamiento. En pacientes que desarrollan letargo de forma inexplicable, o cambios en el estado mental asociado con monoterapia o terapia adjunta con topiramato, se recomienda considerar la encefalopatía hiperamonémica y la cuantificación de los niveles de amonio. Mujeres en edad reproductiva TOPAMAX® puede provocar daño fetal cuando se administra a una mujer embarazada. Hay un riesgo aumentado de parto antes de término y parto prematuro asociado con el uso de MAE, incluyendo topiramato. TOPAMAX® solo debe ser usado durante la gestación si el beneficio potencial justifica el riesgo potencial al feto (ver Restricciones de uso durante el embarazo y la lactancia). Suplementación nutricional.Se puede considerar un suplemento dietético o aumento en la ingesta de alimentos si el paciente está perdiendo peso mientras toma este medicamento. Efectos en la habilidad para manejar y utilizar maquinaria. TOPAMAX® actúa sobre el sistema nervioso central y puede producir somnolencia, mareo u otros síntomas relacionados. También puede producir trastornos visuales y/o visión borrosa. Estos eventos adversos pueden ser potencialmente peligrosos en pacientes que manejan un vehículo u operan maquinaria, particularmente hasta que se establezca la experiencia individual del paciente con el medicamento.
Restricciones de uso durante el embarazo y la lactancia: Embarazo. Estudios en animales han mostrado toxicidad reproductiva (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad - Toxicología reproductiva y del desarrollo). En ratas, topiramato cruza la barrera placentaria. En humanos, topiramato atraviesa la placenta, y concentraciones similares se han reportado en el cordón umbilical y la sangre materna. No existen estudios controlados y adecuados del uso de TOPAMAX® en mujeres embarazadas. TOPAMAX® puede causar daño fetal cuando se administra a una mujer embarazada. Los datos de los registros de embarazo indican que los recién nacidos expuestos a topiramato in utero tienen mayor riesgo de malformaciones congénitas (por ejemplo, defectos craneofaciales como labio y paladar hendido; hipospadias y anormalidades que involucran varios sistemas corporales). Esto se ha reportado con la monoterapia con topiramato y topiramato como parte de un régimen de politerapia. Además, los datos de otros estudios indican que, en comparación con monoterapia, existe un mayor riesgo de efectos teratogénicos asociados con el uso de medicamentos antiepilépticos en terapia de combinación. Se observó riesgo en todas las dosis y los efectos reportados fueron dependientes de la dosis. En mujeres tratadas con topiramato que tuvieron un hijo con una malformación congénita, parece que existe un riesgo mayor de malformaciones en embarazos subsecuentes cuando se exponen a topiramato. Hay un riesgo aumentado de parto antes de término y parto prematuro asociado con el uso de MAE, incluyendo topiramato. En comparación al grupo de referencia que no tomó MAE, los datos de registro para TOPAMAX® como monoterapia muestran una mayor prevalencia de bajo peso al nacer ( < 2500 gramos). Un registro de embarazos reportó un incremento en la frecuencia de recién nacidos que fueron pequeños para la edad gestacional (SGA, por sus siglas en inglés; definido como peso al nacer por debajo del percentil 10 corregido para la edad gestacional, estratificado por sexo) entre aquellos expuestos in utero a topiramato como monoterapia. Se ha observado SGA en t