VOLEBRIS
GSK
Denominacion genérica: Ambrisentan.
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Ambrisentan 5 mg y 10 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: Ambrisentan está indicado para el tratamiento de la hipertensión arterial pulmonar (HAP), para mejorar la habilidad del ejercicio, disminuir los síntomas de la HAP, y retrasar el empeoramiento clínico. En combinación con Tadalafil para reducir el riesgo de falla clínica (un compuesto de muerte, hospitalización por HAP, progresión de la enfermedad y respuesta clínica no satisfactoria) y para aumentar la respuesta clínica satisfactoria y la habilidad para ejercitarse, en pacientes adultos dentro del grupo funcional 1 de la OMS.
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: Se evaluaron los parámetros hemodinámicos invasivos en pacientes con HAP en la línea basal y después de 12 semanas (n = 29) en estudio en Fase 2. El tratamiento con ambrisentan resultó en un incremento significativo en el índice cardíaco promedio (+ 0.3 L/min/m2; 95% CI: 0.15 a 0.51; p < 0.001), y una disminución en la presión arterial pulmonar promedio (-5.2 mmHg; 95% CI: -7.6 a - 2.9; p < 0.001), y resistencia vascular pulmonar promedio (-2.8 mmHg/L/min; 95% CI: -3.8 a -1.8; p < 0.001) para el grupo combinado de ambrisentan. En pacientes con HAP, las reducciones en el péptido natriurético tipo-B (BNP) han demostrado mejorías paralelas en hemodinamia y caminatas de 6 minutos (6MWD). El análisis combinado de los resultados de dos estudios controlados con placebo en Fase 3 demostró que las concentraciones plasmáticas de BNP disminuyeron en pacientes que recibieron ambrisentan por 12 semanas. La concentración plasmática de promedio geométrico de BNP aumentó en 11% en el grupo de placebo, y disminuyó en 29% en el grupo de 2.5 mg, 30% en el grupo de 5 mg, y 45% en el grupo de 10 mg (p < 0.001 para cada grupo de dosis). Se observó una asociación positiva entre el cambio en BNP y la mejoría en la clase de la OMS en la Semana 12. En pacientes con HAP, que recibieron terapia de combinación de primera línea con ambrisentan y Tadalafil, se observó una mayor disminución con respecto a la línea basal del péptido natriurético pro-tipo B N-terminal (NT-pro-BNP) en comparación con monoterapia (disminuciones del porcentaje medio de mínimos cuadrados geométricos de 67% contra 50%, respectivamente; p < 0.0001). Se observaron resultados similares en las comparaciones de la terapia de combinación contra el grupo de monoterapia de ambrisentan (disminución de 56%; p = 0.0111) y el grupo de monoterapia de tadalafil (disminución de 44%; p < 0.0001). La disminución de NT-pro-BNP se observó rápidamente (Semana 4) y se sostuvo hasta la Semana 24. Mecanismo de acción: Ambrisentan es un Antagonista de los Receptores de Endotelina (ARE), selectivo ETA, de la clase ácido propanóico, oralmente activo. La endotelina juega un rol significativo en la fisiopatología de HAP. Ambrisentan bloquea el subtipo de receptor ETA, localizado predominantemente en las células de músculo liso vascular y miocitos cardíacos. Esto previene la activación mediada de endotelina del segundo sistema de mensajería que resulta en proliferación de célula de músculo liso y vasoconstricción. Se espera que la selectividad de ambrisentan para el receptor ETA sobre el receptor ETB retenga la producción mediada del receptor ETB de los vasodilatadores óxido nítrico y prostacilina. Estudios clínicos/Eficacia: Se completaron dos estudios en Fase 3, aleatorizados, doble ciego, controlados con placebo, de seguridad y eficacia multicéntrica de 12 semanas en 393 pacientes con HAP. Los dos estudios fueron idénticos en diseño a excepción de la región geográfica del sitio de investigación y las dosis de ambrisentan. Las dosis seleccionadas para el primer estudio fueron de 5 y 10 mg diariamente (192 sujetos), mientras que el segundo estudio evaluó las dosis de 2.5 y 5 mg diariamente (202 sujetos). El criterio de valoración primario del estudio fue una caminata de 6 minutos (6MWD). Además, fueron evaluados para eficacia el tiempo hasta empeoramiento clínico, clase funcional de la WHO, disnea y Encuesta de Salud SF-36. Se observó un incremento en Caminata de 6 minutos tan pronto como 4 semanas después del inicio del tratamiento con ambrisentan, con una respuesta de dosis observada después de 12 semanas de tratamiento. Los resultados de AMB-321 demostraron que 5 mg y 2.5 mg po qd de ambrisentan mejoraron el 6MWD corregido de placebo en 59.4 metros (p < 0.001) y 32.3 metros (p = 0.022), respectivamente. 2 De manera similar, los resultados de AMB-320 demostraron que 10 mg y 5 mg po qd de ambrisentan mejoraron el 6MWD corregido de placebo en 51.4 metros (p < 0.001) y 30.6 metros (p = 0.008), respectivamente. Se observó una mejoría significativa en 6MWD para cada grupo de dosis de ambrisentan en comparación con placebo, por lo tanto, el análisis pre-especificado de los criterios de valoración secundaria tanto en los estudios controlados con placebo individuales, como en el análisis combinado de estos estudios se enfoca en el grupo de ambrisentan combinado. Los estudios individuales no estuvieron estadísticamente potenciados para examinar los criterios de valoración secundarios. Debido a que el tamaño de muestra mayor del análisis combinado en Fase 3 tuvo el mayor poder de examinar criterios de valoración secundarios, éste proporcionó estimaciones más precisas de estos efectos de tratamiento con ambrisentan. El tiempo hasta el empeoramiento clínico, un indicador de progresión de la enfermedad, fue un criterio de valoración secundario clave en los dos estudios controlados con placebo en Fase 3. La prueba de rango logarítmico para la comparación del grupo de ambrisentan combinado versus placebo demostró que un retraso significativo en el tiempo hasta el empeoramiento clínico de HAP se observó para sujetos que recibieron ambrisentan (p < 0.001). Además, la razón de riesgo fue 0.29 (95% CI: 0.14 a 0.59), indicando un 71% de reducción en la probabilidad de empeoramiento clínico dentro del periodo de tratamiento de 12 semanas para sujetos que recibieron ambrisentan en comparación con placebo. Las conclusiones del análisis combinado fueron apoyadas por tendencias similares en los estudios individuales. En AMB-320, un incremento doble en el número de sujetos con un evento de empeoramiento clínico fue observado en el grupo de placebo en comparación con cada grupo de dosis de ambrisentan; sin embargo, la comparación de rango logarítmico del grupo de ambrisentan combinado versus el grupo de placebo no demostró una diferencia estadísticamente significativa en el tiempo hasta el empeoramiento clínico de HAP (p = 0.214). En AMB-321, un incremento cuádruple en el número de sujetos con un evento de empeoramiento clínico fue observado en el grupo de placebo en comparación con cada grupo de dosis de ambrisentan. La prueba de rango logarítmico demostró un retraso significativo en el tiempo hasta empeoramiento clínico de HAP para la comparación del grupo combinado de ambrisentan versus el grupo de placebo (p < 0.001). El análisis primario de la clase funcional de la OMS utilizó un cambio de 7 puntos desde la escala de la línea basal (+3, +2, +1, 0, -1, -2, -3). Los cambios positivos (+1, +2, ó +3) indicaron un deterioro en la clase funcional de la OMS, y los cambios negativos (-1, -2, ó -3) indicaron una mejoría en la clase funcional de la OMS. Para el análisis combinado, el grupo combinado de ambrisentan demostró una mejoría global estadísticamente significativa en el cambio desde la línea basal en la clase funcional de la OMS en la semana 12 en comparación con placebo (p = 0.009). El efecto de tratamiento positivo observado para el grupo de ambrisentan combinado se debió principalmente a una reducción quíntuple en el porcentaje de sujetos que deterioraron al menos 1 clase de la OMS en comparación con placebo. Se observaron tendencias similares en los análisis individuales de AMB-320 (p = 0.036) 1 y AMB-321 (p = 0.117)2, pero no fueron estadísticamente significativas de acuerdo a los procedimientos estadísticos pre-especificados. Para la Encuesta de Salud SF-36, un análisis de medidas repetidas demostró que la mejoría observada en la escala de funcionamiento físico en el grupo de ambrisentan combinado fue significativamente superior a placebo (p = 0.003). Se observó para cada grupo de dosis de ambrisentan una mejoría en comparación con el grupo de placebo; sin embargo, no hubo respuesta de dosis aparente. Las mejorías comparadas con placebo para el resumen del componente físico general y para las escalas individuales del rol físico, vitalidad y salud general fueron observadas también para el grupo combinado de ambrisentan. El cambio ajustado de placebo desde la línea basal en BDI fue de -0.85 (95% CI: -1.30 a -0.39, p < 0.001) para el grupo de ambrisentan combinado. También se observaron mejorías clínicamente relevantes en BDI en la semana 12 para cada grupo de dosis de ambrisentan en comparación con placebo y estas mejorías parecieron ser mayores para el grupo de 10 mg en comparación con los grupos de 2.5 y 5 mg. Se observó una disminución clínicamente relevante desde la línea basal en BDI para el grupo de ambrisentan combinado en comparación con placebo en AMB-320 y AMB- 321. Esta mejoría fue estadísticamente significativa en AMB-321 (p = -1.1; 0.019), pero debido al procedimiento de comparaciones múltiples pre-especificado, esta mejoría no fue considerada estadísticamente significativa en AMB-320 a pesar de un valor-p pequeño (p = -0.6; 0.017). Los pacientes reclutados en los estudios de Fase 3 fueron elegibles para ser reclutados en un estudio de extensión. El seguimiento a largo plazo de los sujetos que fueron tratados con ambrisentan en los estudios en Fase 3, controlados con placebo y su extensión de marbete abierto (N = 383) muestra que el 93% (95% CI: 90.9 a 95.9) siguió con vida al año (estimación Kaplan- Meier) y 91% (287/314) de los cuales aún tomando ambrisentan se encontraba recibiendo monoterapia con ambrisentan. En 2 años, 85% (95% CI: 81.7 a 88.9) siguieron con vida aún (estimación Kaplan-Meier) y 83% (214/259) de los cuales aún tomando ambrisentan se encontraban recibiendo monoterapia con ambrisentan. A 3 años, 79% (95% CI: 75.2 a 83.4) siguieron con vida aún (estimación Kaplan-Meier) y 79% (147/186) de los cuales aún tomando ambrisentan se encontraban recibiendo monoterapia con ambrisentan. Mejorías desde la línea basal en 6MWD, clase functional OMS, y BDI se mantuvieron con tratamiento a largo plazo por arriba de 3 años en la extensión del estudio a fase 3. Mejorías en 6MWD, clase funcional de la OMS y BDI generalmente se mantuvieron por hasta 3 años en los estudios en Fase 2. La eficacia de ambrisentan pareció similar cuando se administró solo o en combinación con sildenafil y/o un prostanoide, aunque el tamaño de estudio excluyó las comparaciones definitivas del subgrupo. Ambrisentan, en combinación con Tadalafil, para el tratamiento de HAP: El efecto de la terapia de combinación de primera línea con ambrisentan y Tadalafil se demostró en un estudio multicéntrico, doble ciego y controlado con activo, que comparó la combinación de ambrisentan y Tadalafil con la monoterapia de ambrisentan o Tadalafil en pacientes con HAP de clase funcional II- III de la OMS. El estudio reclutó 610 pacientes; 605 pacientes recibieron al menos una dosis del fármaco en estudio y 500 cumplieron los criterios para el análisis primario de eficacia.4 Los pacientes fueron asignados aleatoriamente 2:1:1 a la dosis una vez al día de 10 mg de ambrisentan + 40 mg de Tadalafil, 10 mg de ambrisentan, o 40 mg de Tadalafil. El ambrisentan inició con 5 mg durante 8 semanas y el Tadalafil con 20 mg durante 4 semanas, después de lo cual la dosis se ajustó de forma ascendente si era tolerada. El criterio primario de valoración del estudio fue el tiempo al primer evento de falla clínica, adjudicado por un comité independiente. Además se evaluó el cambio de NT-pro-BNP, el porcentaje de pacientes con respuesta clínica satisfactoria y el cambio de 6MWD desde la basal a la Semana 24. Los pacientes tenían HAP idiopática (53%), HAP hereditaria (3%), o HAP asociada con enfermedades del tejido conectivo, cardiopatías, enfermedad congénita del corazón, infección por VIH o fármacos o toxinas (APAH, 44%). La mediana del tiempo desde el diagnóstico a la primera administración del fármaco del estudio fue de 22 días. Aproximadamente 31% y 69% de los pacientes eran de clase funcional II y III de la OMS, respectivamente. La mediana de la edad de los pacientes fue de 54.4 años (32% fueron ≥65 años). La mayoría de los pacientes eran blancos (90%) y mujeres (78%); 46% eran norteamericanos. Tiempo a la falla clínica: El tiempo a la falla clínica de la HAP fue un criterio de valoración compuesto definido como el tiempo a la primera ocurrencia de muerte (muerte por cualquier causa), hospitalización por HAP agravada, progresión de la enfermedad o respuesta clínica a largo plazo no satisfactoria. La hospitalización por HAP agravada se definió como cualquier hospitalización o HAP agravada, trasplante de pulmón o corazón/pulmón, atrioseptostomía o inicio de terapia parenteral con prostanoides. La progresión de la enfermedad se definió como una disminución > 15% de 6MWD desde la basal, en combinación con síntomas de clase funcional III o IV de la WHO (en dos visitas posbasales consecutivas, separadas por ≥14 días). La respuesta clínica a largo plazo no satisfactoria se definió como cualquier reducción de 6MWD inferior a la basal, combinado con una evaluación de clase funcional III en visitas con 6 meses de separación. Los pacientes tratados con ambrisentan + tadalafil experimentaron una reducción significativa del riesgo de falla clínica contra los pacientes combinados tratados con monoterapia de ambrisentan o tadalafil (p=0.0002), monoterapia de ambrisentan (p=0.0004), o monoterapia de tadalafil (p=0.0045). La reducción del resigo de un evento de falla clínica fue de 50% (HR=0.50, IC de 95%: 0.348, 0.724) con la terapia de combinación, respecto a la monoterapia combinada. Las gráficas de Kaplan-Meier del tiempo a la falla clínica para la terapia de combinación, contra la monoterapia combinada y cada monoterapia, se muestran en la Figura 1; el resumen de los eventos del criterio de valoración primario se muestra en la Tabla 1.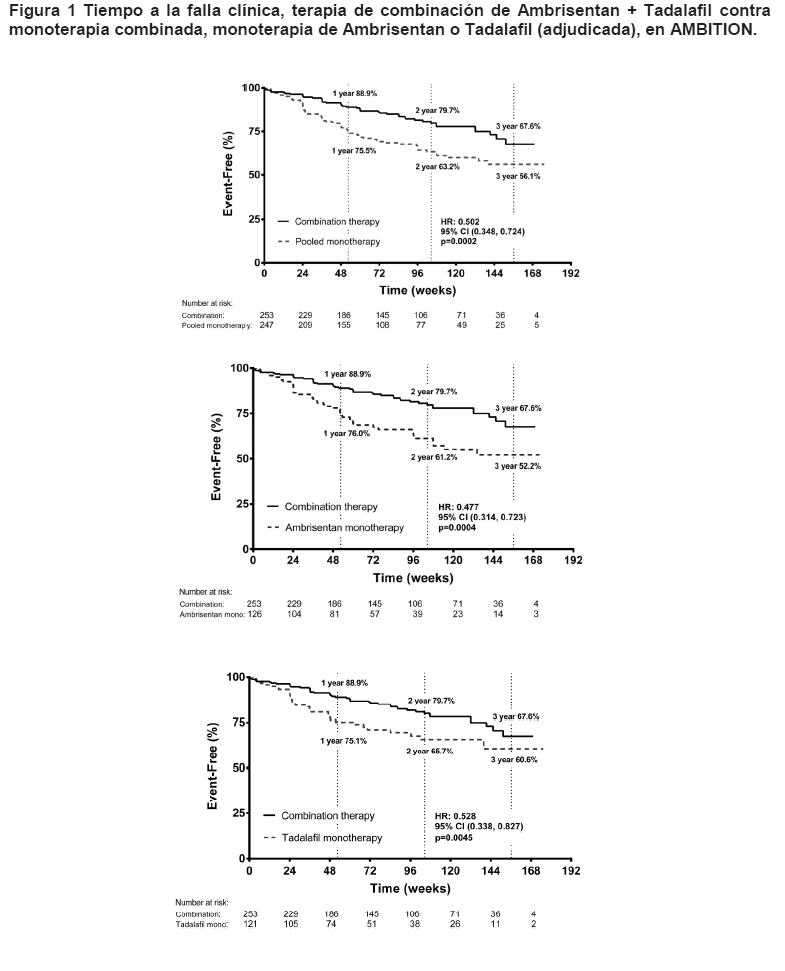
Tiempo desde la asignación aleatoria a la primera falla clínica con estimados de Kaplan-Meier, de las proporciones de las fallas; los valores p que se muestran son las comparaciones log-rango de la terapia de combinación de ambrisentan + tadalafil con la monoterapia combinada y con la monoterapia individual, estratificado por la etiología de la HAP y la clase funcional de la OMS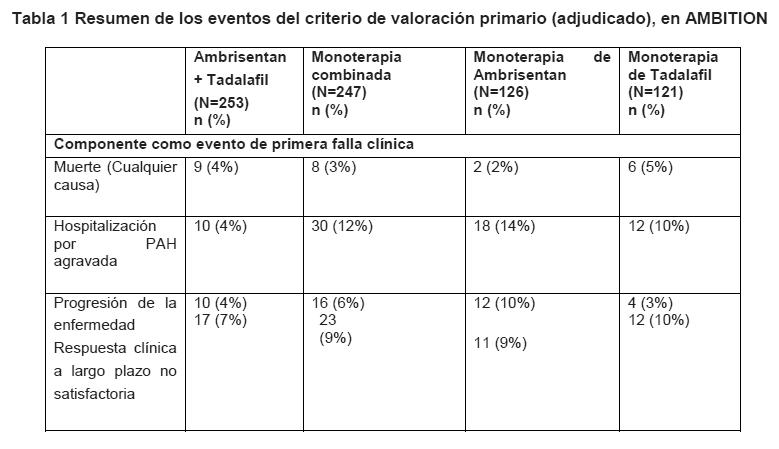
Los resultados de los análisis del tiempo a la falla clínica adjudicada y al primero de cada componente de falla clínica se muestran en la Figura 2.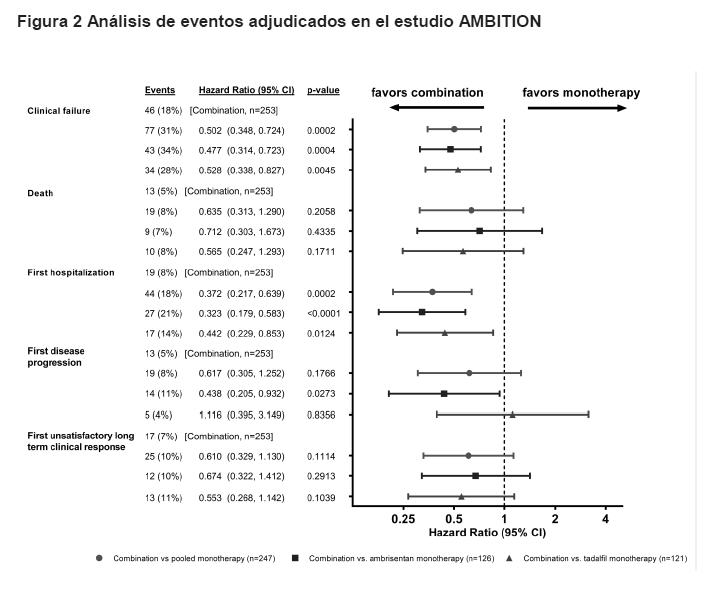
La eficacia del tratamiento de primera línea con ambrisentan + tadalafil sobre el tiempo a la falla clínica se observó en todos los subgrupos (Figura 3).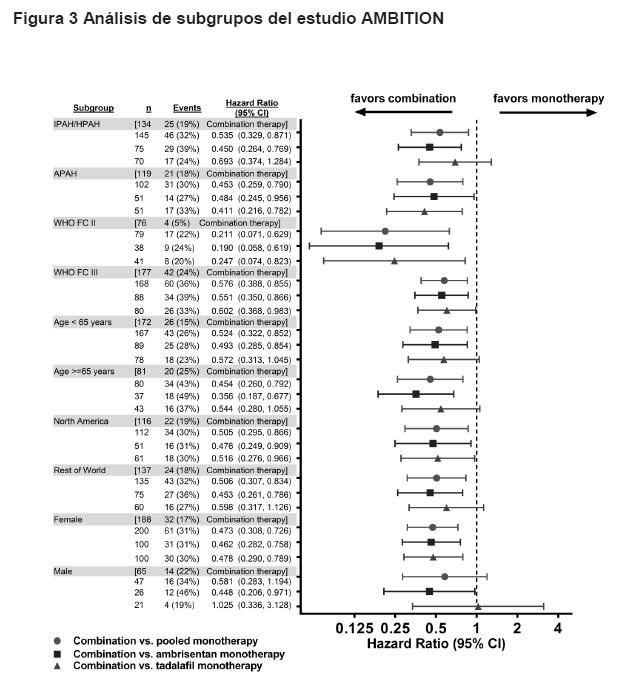
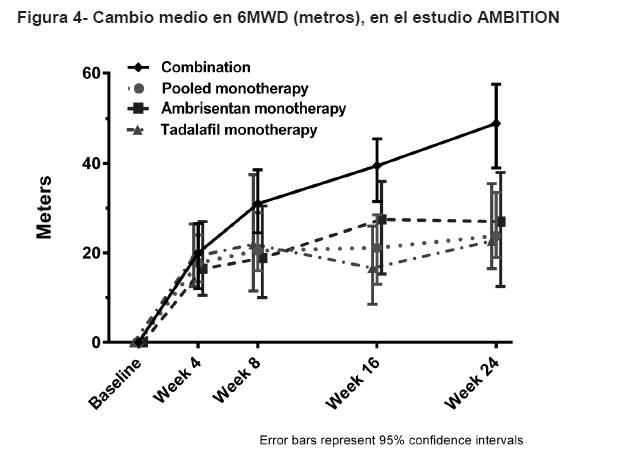
Respuesta clínica: La respuesta clínica satisfactoria a la Semana 24 fue un criterio de valoración secundario compuesto definido como una mejora ≥10% de la C6M, en comparación con la basal, una mejora a o el mantenimiento de los síntomas de clase I o II de la OMS y ningún evento de empeoramiento clínico antes de o en la visita de la Semana 24. El porcentaje de pacientes que lograron una respuesta clínica satisfactoria a la Semana 24 en el grupo de terapia de combinación (39%) fue significativamente mayor que la del grupo combinado de monoterapia (29%, p=0.0264, razón de momios de 1.563, IC de 95%: 1.054, 2.319) y significativamente mayor que en el grupo de monoterapia de tadalafil (27%, p=0.0321, razón de momios de 1.723, IC de 95%: 1.047, 2.833). No se observaron diferencias significativas entre la terapia de combinación y la monoterapia de ambrisentan en la respuesta clínica satisfactoria. Habilidad para ejercitarse: Los resultados de 6MWD a las 24 semanas, para el estudio AMBITION, se muestran en la Tabla 2 y en la Figura 4.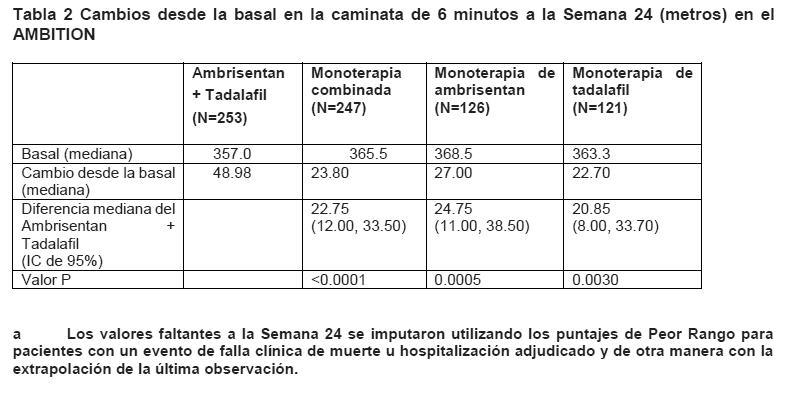
Falta de Beneficio y Aumento en las Hospitalizaciones en la Fibrosis Pulmonar Idiopática: Se realizó un estudio con 492 pacientes (ambrisentan N=329, placebo N=163) con fibrosis pulmonar idiopática (FPI), 11% de los cuales tenían hipertensión arterial pulmonar secundaria (grupo 3 OMS), pero se terminó en fase temprana cuando se determinó que el objetivo primario de eficacia no se alcanzaba. En este estudio, los pacientes fueron aleatorizados a ambrisentan o placebo en razón de 2:1. Se observaron noventa eventos (27%) de progresión de FPI (incluyendo hospitalizaciones por causa respiratoria) o muerte en el grupo con ambrisentan comparado con 28 eventos (17%) en el grupo placebo. La evaluación de los componentes del objetivo primario indicaron que había índices más altos de hospitalizaciones por causa respiratoria, eventos de mortalidad, y disminución de la función respiratoria en el grupo ambrisentan versus placebo. Por lo tanto, Ambrisentan está contraindicado para pacientes con FPI con o sin hipertensión pulmonar secundaria. Propiedades farmacocinéticas: Absorción: Ambrisentan se absorbe rápidamente en humanos. Las concentraciones plasmáticas máximas (Cmax) de ambrisentan ocurren típicamente alrededor de 1.5 horas después de la administración oral tanto en ayunas como sin ayunas. Cmax y el área bajo la curva de tiempo - concentración plasmática (AUC) aumentan proporcionalmente a la dosis sobre el rango de dosis terapéutica. El estado estable generalmente se logra después de 4 días de dosis repetida. Un estudio de efecto alimenticio, en donde se involucró la administración de ambrisentan a voluntarios saludables bajo condiciones de ayuno y con comida rica en grasas, indicó que la Cmax disminuyó en un 12% mientras que el AUC permaneció sin cambios. Esta disminución en la concentración del pico no es clínicamente significativa, y por tanto ambrisentan puede ser administrado con o sin comida. Distribución: Ambrisentan se enlaza altamente a proteína plasmática. El enlace a proteína plasmática in vitro de ambrisentan fue, en promedio, 98.8% e independiente de la concentración sobre el rango de 0.2 - 20 microgramos/mL. Ambrisentan está principalmente enlazado a albúmina (96.5%) y en menor medida a a 1-ácido glicoproteína. La distribución de ambrisentan en los glóbulos rojos es baja, con una proporción promedio de sangre: plasma de 0.57 y 0.61 en hombres y mujeres, respectivamente. Metabolismo: Ambrisentan se glucuroniza mediante varias enzimas UGT (UGT1A9S, UGT2B7S y UGT1A3S) para formar glucuronido de ambrisentan7. Ambrisentan se somete también a metabolismos oxidativos, principalmente mediante CPY3A4 y en menor medida mediante CYP3A5 y CYP2C19, para formar 4-hidroximetil ambrisentan, el cual se glucuroniza adicionalmente a 4-hidroximetil ambrisentan glucuronida. En plasma, el AUC de 4-hidroximetil ambrisentan representa aproximadamente el 4% relativo al AUC de ambrisentan original. Además, la afinidad de enlace de 4-hidroxymetil ambrisentan para el receptor ETA humano es más de 100 veces menor que ambrisentan. Por lo tanto, no se espera que 4-hidroxymetil ambrisentan contribuya a la actividad farmacológica de ambrisentan. Los estudios in vitro que utilizaron cultivo de hepatocito humano y de rata han demostrado que ambrisentan es un sustrato posible para el transportador de afluencia hepática OATP y el transportador de eflujo P-gp, pero no para la afluencia hepática o eflujo de proteína co- transportadora de taurocolato de sodio (NTCP) o bomba exportadora de sal biliar (BSEP), respectivamente. Los datos in vitro indican que ambrisentan no muestra una inhibición marcada de UGT1A1, UGT1A6, UGT1A9, UGT2B7 o las enzimas del citocromo 2C19, 3A4 a concentraciones de hasta 300 mM. Además, los estudios in vitro que usan líneas celulares transfectadas con los genes transportadores humanos mostraron que ambrisentan no inhibe (P-gp), (BCRP) o (BSEP) a concentraciones de hasta 100 mM.. Ambrisentan mostró inhibición in vitro débil de OATP1B1, OATP1B3 y cotransportador de taurocolato sódico (NTCP) con valores IC50 de 47 mM, 45 mM, y aproximadamente 100 mM 45. Estudios in vitro en hepatocitos de rata y humanos no mostraron evidencia de inhibición de ambrisentan de NTCP, OATP, BSEP. Asimismo, ambrisentan no induce la expresión proteínica de P-pg o BSEP en hepatocitos de rata46. Tomados en conjunto, los datos in vitro sugieren que no sería de esperarse que ambrisentan, a concentraciones clínicamente relevantes,tuviera un efecto sobre UGT o enzimas del citocromo 2C19, 3A4 o transporte vía BSEP, BCRP, P-gp, OATP1B1/3, o NTCP. En voluntarios sanos, se estudiaron los efectos producidos por la dosificación repetida de ciclosporina A (100 - 150 mg dos veces al día) en el perfil farmacocinético en estado estacionario del ambrisentan (5 mg una vez al día), así como los efectos producidos por la dosificación repetida de ambrisentan (5 mg una vez al día) en el perfil farmacocinético en estado estacionario de la ciclosporina A (100 - 150 mg dos veces al día). La Cmax y el AUC(0-t) del ambrisentan experimentaron un incremento (48% y 121%, respectivamente) en presencia de múltiples dosis de ciclosporina A. Con base en estos cambios, la dosis de ambrisentan debe limitarse a 5 mg una vez al día, cuando se coadministre con ciclosporina A (véase Posología y Método de Administración). Sin embargo, la administración de dosis múltiples de ambrisentan no tuvo efectos clínicamente relevantes en el nivel de exposición a la ciclosporina A, por lo cual no es necesario ajustar la dosis de ciclosporina A. Se investigó en 16 voluntarios saludables los efectos de la dosis repetida de ketoconazol (400 mg una vez al día) en la farmacocinética de dosis única de ambrisentan 10 mg. Las exposiciones de ambrisentan según se midió mediante AUC(0-inf) y Cmax se incrementaron en 35% y 20%, respectivamente. Es improbable que este cambio en la exposición tenga alguna relevancia clínica y por lo tanto ambrisentan puede ser coadministrado con ketoconazol. Con base en los resultados de este estudio, no se requiere ajustar la dosis de ambrisentan cuando éste se coadministra con inhibidores de la CYP3A. En voluntarios sanos, se estudiaron los efectos producidos por la dosificación repetida y aguda de rifampina (600 mg una vez al día) en el perfil farmacocinético en estado estacionario del ambrisentan (10 mg una vez al día). Después de la administración de dosis iniciales de rifampina, se observó un incremento transitorio en el AUC(0-t) de ambrisentan (87% y 79% después de la primera y la segunda dosis de rifampina, respectivamente). Sin embargo, por el día 7, no hubo efectos clínicamente relevantes en el nivel de exposición al ambrisentan, después de la administración de dosis múltiples de rifampina. No se requiere ajustar la dosis de ambrisentan cuando éste se coadministra con rifampina. En voluntarios sanos, se estudio el efecto de ritonavir en el estado de equilibrio farmacocinético de ambrisentan, y el efecto de ambrisentan en el estado de equilibrio farmacocinético de ritonavir. La co-administración de ritonavir (100 mg una vez al día) con ambrisentan (5 mg una vez al día) durante diez días resultó en un ligero cambio en ambrisentan, con un 7% de aumento en Cmax y un 5% de disminución en AUC(0-t) de ambrisentan. No se observe un cambio significativo en ritonavir (Cmax o AUC(0-t)) cuando se co-administró con ambrisentan. Basado en estos resultados, ritonavir no tuvo efecto significativo en la farmacocinética de ambrisentan, y ambrisentan no tuvo efecto significativo en la farmacocinética de ritonavir a las dosis probadas. En voluntarios sanos, se estudio el efecto de tacrolimus en el estado de equilibrio farmacocinético de ambrisentan. La co-administración de tacrolimus (a una dosis oral de 0.05 mg/kg dos veces al día) con ambrisentan (5 mg una vez al día) durante seis días resultó en una disminución de 3% en Cmax y sin cambios en AUC(0-t) de ambrisentan, indicando que tacrolimus no tiene efecto en la farmacocinética de ambrisentan. En voluntarios sanos, se estudio el efecto de micofenolato mofetilo (MMF) en el estado de equilibrio farmacocinético de ambrisentan, y el efecto de ambrisentan en el estado de equilibrio farmacocinético de los metabolitos de MMF ácido micofenólico (MPA) y glucoronido del ácido micofenólico (MPAG). La co-administration de MMF (1000 mg dos veces al día) con ambrisentan (5 mg una vez al día) durante cinco días resultó en una ligera disminución en ambrisentan, con una disminución de 8% y 4% en Cmax y AUC(0-t) de ambrisentan, respectivamente. No se observaron cambios significativos en los parámetros farmacocinéticos en estado de equilibrio de MPA o MPAG (Cmax o AUC(0-t)) cuando MMF se co-administró con ambrisentan, con la excepción de un aumento estadísticamente significativo de 14% en Cmax de MPA. Este pequeño aumento estadísticamente significativo no se consideró como clínicamente relevante. Basado en estos resultados, MMF no tiene efecto significativo en la farmacocinética de ambrisentan, y ambrisentan no tiene efecto clínicamente relevante en la farmacocinética de los metabolitos MPA o MPAG de MMF. Se investigó en 19 voluntarios saludables el efecto de dosis de 7 días de sildenafil (20 mg tres veces al día) sobre la farmacocinética de una dosis única de ambrisentan, y los efectos de dosis de 7 días de ambrisentan (10 mg una vez al día) en la farmacocinética de una dosis única de sildenafil. No hubo ningún otro cambio en los parámetros farmacocinéticos de sildenafil, N-desmetil-sildenafil y ambrisentan, con excepción de un incremento del 13% en Cmax de sildenafil después de la coadministración con ambrisentan. Este ligero aumento en Cmax de sildenafil no se considera clínicamente relevante. La administración concomitante de una dosis única de ambrisentan (10 mg) no tuvo efectos clínicamente relevantes en la farmacocinética de ambrisentan ni en su metabolito, 4 hidroximetil ambrisentan en voluntarios saludables que recibieron tadalafil (40 mg una vez al día). De manera similar, la farmacocinética de dosis única de tadalafil (40 mg) no estuvo afectada por dosis múltiples de ambrisentan (10 mg una vez al día). Se estudió en mujeres voluntarias saludables el efecto de dosis de 12 días con ambrisentan (10 mg una vez al día) sobre la farmacocinética de una dosis única de anticonceptivo oral con contenido de 1 mg de noretindrona y 35 microgramos de etinil estradiol. La Cmax y el AUC(0-∞) disminuyeron ligeramente para etinil estradiol (8% y 4%, respectivamente), y aumentaron ligeramente para noretindrona (13% y 14%, respectivamente). Estos cambios en la exposición a etinil estradiol o noretindrona fueron pequeños y es improbable que sean clínicamente significativos. Se investigó en 20 voluntarios saludables los efectos de ambrisentan en estado estable (10 mg una vez al día) sobre la farmacocinética y farmacodinamia de una dosis única de warfarina (25 mg), según se midió mediante tiempo de protrombina (PT) y relación normalizada internacional (INR). Ambrisentan no tuvo ningún efecto clínicamente relevante sobre la farmacocinética o la farmacodinamia de warfarina. De manera similar, la coadministración con warfarina no afecta la farmacocinética de ambrisentan. Se estudió en 15 voluntarios saludables el efecto de dosis repetida de ambrisentan (10 mg) sobre la farmacocinética de digoxina de dosis única. Las dosis múltiples de ambrisentan resultaron en ligeros aumentos en AUC0-última de digoxina y concentraciones mínimas, e incremento de 29% en Cmax de digoxina. El incremento en la exposición de digoxina observado en presencia de dosis múltiples de ambrisentan no se considera clínicamente relevante, y ningún ajuste de dosis de ambrisentan estará garantizado. Eliminación: Ambrisentan y sus metabolitos son eliminados principalmente en la bilis después del metabolismo extra-hepático y/o hepático. En las heces, el 40% de la dosis se recupera como ambrisentan original y el 21% como el 4-hidroximetil ambrisentan. Aproximadamente el 22% de la dosis administrada se recupera en la orina después de la administración oral, siendo que el 3.3% es ambrisentan sin cambios y la parte restante aparece como metabolitos glucurónidos. La vida media de eliminación plasmática en estado estable cayó en un rango de 13.6 a 16.5 horas en voluntarios saludables y de 12.9 a 17.9 horas en pacientes con HAP. Farmacocinética en poblaciones especiales Edad y Género: Basándose en los resultados de un análisis farmacocinético de la población en voluntarios saludables y pacientes con PAH, la farmacocinética de ambrisentan no estuvo significativamente influenciada por la edad o el género (véase Dosis y vía de Administración). Insuficiencia Hepática: La farmacocinética de ambrisentan no ha sido estudiada en sujetos con deterioro hepático severo o con transaminasas hepáticas elevadas clínicamente significativas. Sin embargo, se espera que el deterioro hepático incremente la exposición (Cmax y AUC) a ambrisentan, ya que sus rutas principales de metabolismo son glucuronidación y, en menor medida mediante oxidación, con eliminación subsecuente en la bilis. No se ha evaluado la magnitud de este efecto ni ningún impacto sobre la seguridad y eficacia. Por lo tanto, no se recomienda ambrisentan en esta población de pacientes. Basándose en un modelo farmacocinético de población final desarrollado, basado en datos farmacocinéticos de sujetos en estudio clínico que reciben ambrisentan, hubo una relación significativa entre ambrisentan CL/F y la función hepática según se evaluó mediante bilirrubina total. Sin embargo, las magnitudes del cambio en la bilirrubina total fueron relativamente pequeñas. Insuficiencia Renal: No se ha estudiado la farmacocinética de ambrisentan en sujetos con deterioro renal. Sin embargo, el metabolismo renal y la excreción de ambrisentan son mínimas, así que es improbable que el deterioro renal incremente significativamente la exposición a ambrisentan. Basándose en un modelo farmacocinético de población final desarrollado, basado en datos farmacocinéticos de sujetos en estudio clínico que reciben ambrisentan, hubo una relación significativa entre ambrisentan CL/F y la función renal según se evaluó mediante la eliminación de la creatinina (Clcr). Sin embargo, las magnitudes del cambio en la depuración de ambrisentan fueron relativamente modestas, así que es improbable que sean de relevancia clínica.
Contraindicaciones: Ambrisentan está contraindicado en mujeres embarazadas. En los pacientes con hipersensibilidad al ambrisentan y/o a los componentes de la fórmula. Ambrisentan está contraindicado en fibrosis pulmonar idiopática (IPF) con o sin hipertensión pulmonar secundaria.
Precauciones generales: Insuficiencia hepática: Se han observado elevaciones de las enzimas hepáticas con Antagonista de los Receptores de Endotelina (ARE). Realizar pruebas de funcionamiento hepático de acuerdo a la clínica.Si las aminotransferasas (alanina aminotransferasa, ALT o aspartato aminotransferasa, AST) son mayores a 3 veces el límite superior del nivel normal, no se recomienda el inicio de ambrisentan. Los pacientes con insuficiencia cardiaca derecha clínicamente significativa, enfermedad hepática pre-existente, elevaciones previas de aminotransferasas debidas a medicamentos o tomando concurrentemente medicamentos concomitantemente que se sabe aumentan las aminotransferasas pueden estar en un riesgo mayor de desarrollar elevación de aminotransferasas con ambrisentan. Se debe vigilar las aminotransferasas según lo indique el estado clínico. Si el paciente desarrolla elevaciones de aminotransferasa clínicamente significativas o si las elevaciones de éstas están acompañadas por signos o síntomas de lesión hepática (por ejemplo, ictericia), la terapia con ambrisentan deberá ser descontinuada. En pacientes sin síntomas clínicos de lesión hepática o de ictericia debe ser considerado el reinicio de ambrisentan después de la resolución de las anormalidades de enzimas hepáticas. Se sabe que ocurre daño hepático y hepatitis autoinmune en pacientes con HAP y frecuentemente se han encontrado autoanticuerpos en HAPI (HAP Idiopática). Casos consistentes con hepatitis autoinmune, incluyendo la posible exacerbación de hepatitis autoinmune subyacente, y se ha reportado daño hepático con la terapia de ambrisentan, aunque aún no está clara la contribución de ambrisentan en esos eventos. Por lo tanto, se debe monitorear a los pacientes para signos de daño hepático y se debe tener precaución cuando se use ambrisentan solo o concomitantemente con otros medicamentos que se sepa están asociados a daño debido a que no se sabe si hay un efecto aditivo de ambrisentan con esos agentes. Debe optimizarse el manejo de la hepatitis autoinmune en pacientes con HAP previamente al inicio y durante el tratamiento con ambrisentan. Se debe descontinuar ambrisentan si los pacientes desarrollan signos o síntomas de hepatitis, o sufren exacerbación de hepatitis autoinmune existente. Otros ERAs se han asociado con elevaciones de aminotransferasea (AST, ALT), hepatotoxicidad, y casos de insuficiencia hepática (véase Efectos Indeseables). En pacientes que desarrollen insuficiencia hepatica después del inicio de la terapia con ambrisentan, se debe investigar a fondo la causa del daño hepático. Se debe descontinuar el ambrisentan si las elevaciones de aminotransferasas hepaticas son > 5x ULN o si las elevaciones se acompañan de bilirrubina > 2x ULN, o por signos o síntomas de disfunción hepática o se excluyen otras causas. Cambios Hematológicos: Se han observado reducciones en concentraciones de hemoglobina y hematocritos con ERA incluyendo ambrisentan, y ha habido casos en donde esto ha resultado en anemia, requiriendo transfusión algunas veces. Se observaron disminuciones de hemoglobina y hematocrito en los estudios clínicos dentro de las primeras semanas de terapia y generalmente se estabilizaron posteriormente. La disminución promedio en hemoglobina desde la línea basal hasta el final del tratamiento fue de 0.8 g/dl para pacientes que están recibieron ambrisentan en los estudios controlados con placebo de 12 semanas. Las disminuciones promedio a partir de la basal (variando desde 0.9 a 1.2 g/dL) en las concentraciones de hemoglobina persistieron durante los 4 años de tratamiento con ambrisentan en la extensión a largo plazo de los estudios clínicos piloto Fase 3. Se recomienda que se mida la hemoglobina antes del inicio de ambrisentan, de nuevo al mes y posteriormente de forma periódica. El inicio de la terapia con ambrisentan no se recomienda en pacientes con anemia clínicamente significativa. En caso de que se observe una disminución clínicamente significativa en hemoglobina durante la terapia y se hayan excluido otras causas, deberá considerarse la descontinuación de ambrisentan. Retención de Líquidos: Se ha observado edema periférico con ERA, incluyendo ambrisentan. El edema periférico puede ser también una consecuencia clínica de PAH. La mayoría de los casos de edema periférico en estudios clínicos con ambrisentan fueron de leves a moderados en severidad. Se han recibido reportes post-comercialización de retención de líquidos ocurridos dentro de unas semanas después de comenzar ambrisentan, y en algunos casos, han requerido la intervención de un diurético o de hospitalización por manejo de fluidos o insuficiencia cardíaca descompensada. Si los pacientes tienen sobrecarga de líquidos pre-existente, esto deberá ser manejado de manera clínicamente apropiada antes de comenzar con ambrisentan. Si durante la terapia con ambrisentan se desarrolla retención de líquidos clínicamente significativa, con o sin aumento de peso asociado, deberán tomarse nuevas evaluaciones para determinar la causa, tales como insuficiencia cardíaca subyacente o ambrisentan, y la posible necesidad para tratamiento específico o descontinuación de terapia con ambrisentan. Enfermedad Pulmonar Venooclusiva: Si los pacientes desarrollan edema pulmonar agudo durante el inicio de la terapia con agentes vasodilatadores, como algún antagonista de los receptores endoteliales, se deberá contemplar la posibilidad de que desarrollen enfermedad pulmonar veno-oclusiva. Efectos sobre la capacidad para conducir y el manejo de maquinaria: No ha habido estudios que investiguen el efecto de ambrisentan sobre la capacidad para conducir y la habilidad para operar maquinaria. Un efecto perjudicial en dichas actividades no está previsto desde el perfil de seguridad conocido.
Restricciones de uso durante el embarazo y la lactancia: Ambrisentan está contraindicado en el embarazo. Estudios animales en ratas y conejos han demostrado que ambrisentan es teratogénico. La teratogenicidad es un efecto de clase de los ERA. Las mujeres con potencial reproductivo deberán ser advertidas del riesgo de daño fetal si se toma ambrisentan durante el embarazo. Debe de excluirse el embarazo antes del inicio del tratamiento con ambrisentan y prevenirse posteriormente con anticonceptivos fiables. Se recomienda como clínicamente indicado la prueba de embarazo antes de iniciar el tratamiento con ambrisentan. En caso de embarazo o sospecha del mismo, las mujeres con potencial reproductivo deben ser aconsejadas para que contacten inmediatamente a su médico tratante. Lactancia: No se sabe si ambrisentan se excreta en la leche humana. Ambrisentan no se recomienda para uso en madres lactantes. Fertilidad: El desarrollo de atrofia tubular testicular en animales macho ha sido vinculado a la administración crónica de ERA, incluyendo ambrisentan. El efecto sobre la fertilidad humana masculina no se conoce.
Reacciones secundarias y adversas: Experiencia de Estudios Clínicos Pivotales: La seguridad de ambrisentan fue evaluada durante estudios clínicos en más de 480 pacientes con HAP. A continuación se listan por clase de sistema-órgano y frecuencia las reacciones farmacológicas adversas (ADR) de los datos de los estudios clínicos. Las frecuencias están ajustadas para considerar el placebo se definen como comunes (≥ 1/100, < 1/10) y no comunes (≥ 1/1000, < 1/100). Puede ser que las categorías de frecuencia de reacciones adversas asignadas basadas en la experiencia de estudios clínicos no reflejen la frecuencia de los eventos adversos que ocurren durante la práctica clínica normal. Trastornos de la sangre y del sistema linfático: Común: Anemia (disminución en hemoglobina y/o hematocrito). Trastornos del sistema inmune: No común: Hipersensibilidad (por ejemplo, angioedema, erupción). Trastornos del sistema nervioso: Común: Cefalea. Trastornos cardíacos: Común: Palpitaciones. Trastornos vasculares: Común: Ruborización. Trastornos respiratorios, torácicos y mediastinales: Común: Congestión nasal, sinusitis, nasofaringitis. La incidencia de congestión nasal estuvo relaci