CAMPTOSAR®
PFIZER
Denominación genérica: Irinotecan.
Forma farmacéutica y formulación: Solución Inyectable. Cada vial contiene: Clorhidrato de Irinotecan 40 mg, 100 mg y 300 mg. Excipientes cbp 2 mL, 5 mL y 15 mL.
Indicaciones terapéuticas: El irinotecan está indicado como tratamiento de agente único o combinación, para pacientes con: Carcinoma metastásico de colon o recto, que haya recurrido o progresado después de una terapia a base de 5-fluorouracilo (5-FU). Carcinoma metastásico de colon o recto, sin tratamiento previo. Cáncer pulmonar de células no pequeñas. Cáncer pulmonar de células pequeñas. Cáncer cervical. Cáncer ovárico. Cáncer gástrico inoperable o recurrente. Cáncer esofágico. El Irinotecan en combinación con Cetuximab está indicado para el tratamiento de pacientes con cáncer colorrectal metastásico que expresen el factor de Crecimiento Epidérmico (EGFR) KRAS del tipo salvaje, que no han recibido tratamiento previo para la enfermedad metastásica o después de falla a terapia citotóxica que incluya Irinotecan (Ver sección Propiedades farmacocinéticas y farmacodinámicas). El irinotecan en combinación con 5-FU, ácido folínico (AF) y bevacizumab está indicado para el tratamiento de primera línea de pacientes con carcinoma metastásico de colon o recto. (Ver sección Propiedades farmacocinéticas y farmacodinámicas). El Irinotecan en combinación con capecitabine con o sin bevacizumab está indicado para el tratamiento de primera línea en pacientes con carcinoma metastásico colorrectal (Ver sección Propiedades farmacocinéticas y farmacodinámicas). El irinotecan está indicado como tratamiento de agente único, en pacientes con: Cáncer de mama inoperable o recurrente. Carcinoma de Células escamosas de piel. Melanoma maligno. Linfoma maligno. Cáncer pancreático. Glioma.
Farmacocinética y farmacodinamia: Propiedades Farmacocinéticas: Absorción y Distribución: Después de una infusión intravenosa en humanos, las concentraciones en plasma del irinotecan declinan en forma multiexponencial, con una vida media de eliminación terminal de aproximadamente 6 horas. La vida media de eliminación terminal del metabolito activo SN-38 es de aproximadamente 10 horas. Las vidas medias de las formas de lactona (activas) del irinotecan y del SN-38, son similares a las del irinotecan y SN-38 total, ya que las formas lactonas e hidroxiácidos se encuentran en equilibrio. Sobre el intervalo de dosis de 50 a 350 mg/m2, el ABC del irinotecan aumenta linealmente con la dosis; el ABC del SN-38 se incrementa menos que proporcionalmente con la dosis. Las concentraciones máximas del metabolito activo SN-38, se observan generalmente después de una hora de la finalización de una infusión de 90 minutos de irinotecan. El irinotecan exhibe una unión moderada a las proteínas del plasma (30-68%); mientras que el SN-38 tiene un alto grado de unión a las proteínas plasmáticas humanas (aproximadamente 95%). La albúmina es la proteína plasmática a la cual se unen predominantemente el irinotecan y el SN-38. Metabolismo y Excreción: La conversión metabólica del irinotecan al metabolito activo SN-38 está mediada por enzimas carboxilesterasas y ocurre primariamente en el hígado. El SN-38 experimenta subsecuentemente conjugación en mayor medida por la enzima UDP-glucuronosil transferasa 1A1 (UGT1A1) para formar un metabolito glucurónido. La actividad de UGT1A1 es reducida en personas con polimorfismos genéticos que conllevan a la reducción de la actividad enzimática como la del polimorfismo UGT1A1*28. (Ver sección Precauciones generales). El glucurónido del SN-38 tuvo un 1/50 a 1/100 de la actividad del SN-38, en ensayos de citotoxicidad empleando dos líneas celulares in vitro. La disposición del irinotecan en humanos, no ha sido aclarada completamente. La excreción urinaria del irinotecan es del 11% al 20%; la del SN-38 < 1% y la del glucurónido del SN-38 3%. La excreción acumulativa biliar y urinaria de irinotecan y sus metabolitos (SN-38 y glucurónido del SN-38) durante un periodo de 48 horas después de la administración de irinotecan en dos pacientes, estuvo en el rango de aproximadamente 25% (100 mg/m2) a 50% (300 mg/m2). Farmacocinética en Poblaciones Especiales: Geriátrica. La farmacocinética el irinotecan administrado usando el régimen semanal, fue evaluada en un estudio de 183 pacientes que se diseñó prospectivamente para investigar el efecto de la edad sobre la toxicidad del irinotecan. Los resultados de este estudio indican que no hay diferencias en la farmacocinética del irinotecan, el SN-38 y el glucurónido del SN-38, entre los pacientes con ≥65 años de edad y los de < 65 años de edad. En un estudio de 162 pacientes que no se diseñó prospectivamente para investigar el efecto de la edad, se evidenciaron pequeñas diferencias, pero estadísticamente significativas, en los parámetros farmacocinéticos normalizados por la dosis, entre los pacientes con ≥65 años de edad y los de < 65 años de edad. Aunque el valor de AUC0-24 normalizada por la dosis del SN-38 en pacientes de ≥65 años de edad, fue 11% mayor que la de los pacientes < 65 años de edad, esta diferencia no fue estadísticamente significativa. Pediátrica. (Ver la sección Precauciones Generales - Poblaciones Especiales: Pediátrica). La farmacocinética del irinotecan y sus principales metabolitos en niños se investigó en estudios clínicos realizados en EU y Europa. En general, los resultados y las conclusiones sobre la farmacocinética del irinotecan fueron comparables en los estudios estadounidenses y europeos. Las diferencias que surjan entre estos estudios probablemente se pueden atribuir a las diferencias en las dosis investigadas (20 a 200 mg/m2 y 200 a 720 mg/m2 en los estudios estadounidenses y europeos, respectivamente) y la marcada variabilidad entre pacientes en los valores determinados para los parámetros farmacocinéticos del irinotecan y el SN-38. Estudios en Estados Unidos. Los parámetros farmacocinéticos del irinotecan y del SN-38, fueron determinados en 2 estudios pediátricos de tumores sólidos, en niveles de dosis de 50 mg/m2 (infusión de 60-min, n=48) y 125 mg/m2 (infusión de 90-min, n=6). La depuración del irinotecan (media ± D.E.) fue 17,3 ± 6,7 L/h/m2, para la dosis de 50 mg/m2 y 16,2 ± 4,6 L/h/m2 para la dosis de 125 mg/m2, que son comparables a las de los adultos. Los valores normalizados del AUC del SN-38 de los adultos y niños fueron comparables. En niños se observó una acumulación mínima del irinotecan y del SN-38, con regímenes de dosificación diarios [diariamente x 5 cada 3 semanas ó (diariamente x 5) x 2 semanas, cada 3 semanas]. Un hallazgo de que los valores de AUC de SN-38 normalizados por las dosis fueron comparables entre adultos y niños no guardó concordancia con el incremento de la depuración del irinotecan observada en los niños y quizá reflejó la marcada variabilidad entre pacientes (los valores %CV para el ABC de SN-38 fueron de 84 a 120%). En verdad, la exposición a SN-38 en niños fue aproximadamente 30% más baja que en adultos cuando se hizo la comparación sin tener en cuenta la variabilidad de los datos. Estudios Europeos: La farmacocinética del irinotecan y sus principales metabolitos se investigó en niños con tumores sólidos en un estudio de Fase I en niveles de dosis de 200 a 720 mg/m2 (infusión de 2 horas, n = 77). La exposición sistémica al irinotecan, SN-38, APC, y NPC fue proporcional a la dosis. Los parámetros farmacocinéticos del irinotecan y sus metabolitos demostraron una marcada variabilidad entre pacientes con valores (media ± DE) de depuración plasmática del irinotecan de 18 ± 8 L/h/m2 y volumen de distribución en el estado de equilibrio de 104 ± 84 L/m2. La depuración del irinotecan fue 26% más baja en adolescentes que en niños y las exposiciones a SN-38 y SN-38G normalizadas por dosis fueron de 52% y 105% más altas en adolescentes que en niños, respectivamente. La depuración del irinotecan fue más alta y los valores normalizados para las dosis de la exposición a SN-38, SN-38G y APC fueron más bajos en los niños que en los adultos. Se llevó a cabo un análisis de la farmacocinética poblacional del irinotecan en 83 niños y adolescentes con casos en recaída o refractarios de rabdomiosarcoma, tumor neuroectodérmico primitivo (PNET) incluido el meduloblastoma o el neuroblastoma que recibieron 600 mg/ m2 de irinotecan en infusión en una hora una vez cada 3 semanas como parte de un estudio de fase II. Los valores medios de la depuración y el AUC del irinotecan mostraron gran variabilidad inter e intra-individuos y fueron similares a los que se determinaron con la misma dosis en el estudio europeo de fase I en niños. Género. Aparentemente, la farmacocinética del irinotecan no es influida por el género. Raza. No se ha evaluado la influencia de la raza sobre la farmacocinética del irinotecan. Insuficiencia Hepática. (Ver la sección Dosis y Vía de Administración - Poblaciones Especiales). La depuración del irinotecan está disminuida en pacientes con disfunción hepática, mientras que la exposición relativa al metabolito activo SN-38 está aumentada. Las magnitudes de esos efectos son proporcionales al grado de deterioro hepático, medidos por los aumentos en las concentraciones séricas de bilirrubina total y transaminasas. Insuficiencia Renal. La influencia de la insuficiencia renal sobre la farmacocinética del irinotecan, no ha sido evaluada (Ver la sección Dosis y Vía de Administración - Pacientes con la Función Renal Deteriorada). Propiedades Farmacodinámicas: Clase Terapéutica: El clorhidrato de Irinotecan es un agente antineoplásico de la clase de los inhibidores de la topoisomerasa I, investigado clínicamente como CPT-11. El irinotecan es un derivado semisintético de la campotecina, un alcaloide extraído de plantas como la Campotheca acuminata o químicamente sintetizado. Mecanismo de Acción: El irinotecan y su metabolito activo, el SN-38, se unen al complejo topoisomerasa I-ADN y previenen que se unan de nuevo las cadenas simples divididas. Investigaciones actuales sugieren que la citotoxicidad de irinotecan se debe al daño que se produce en la doble hélice del DNA durante la síntesis del mismo, cuando las enzimas de replicación interactúan con el complejo ternario formado por la topoisomerasa I, el DNA y el irinotecan o SN-38. El Irinotecan sirve como un precursor soluble en agua del metabolito lipofílico SN-38. El SN-38 se forma a partir de la ruptura del enlace carbamato entre la porción de camptotecina y la cadena lateral dipiperidino, -mediado por la carboxiesteresa-. El SN-38 es aproximadamente 1,000 veces más potente que el irinotecan inhibiendo la topoisomerasa I purificada de líneas celulares de tumores humanos y roedores. Los ensayos de citotoxicidad in vitro, muestran que la potencia del SN-38 varía de 2 a 2,000 veces, en relación a la del irinotecan. Sin embargo, los valores del área bajo la curva concentración plasmática versus tiempo (ABC) del SN-38, son de 2% a 8% los del irinotecan y el SN-38 se encuentra unido en un 95% a las proteínas plasmáticas, en comparación con una unión a las proteínas plasmáticas de aproximadamente 50% del irinotecan. Todavía se desconoce cuál es la contribución precisa del SN-38 a la actividad del irinotecan. Ambos, el irinotecan y el SN-38, existen bajo las formas de la lactona activa y del anión hidroxiácido inactiva. Entre las dos formas existe un equilibrio dependiente del pH, de manera que el pH ácido promueve la formación de la lactona, mientras que un pH más básico favorece la forma del anión hidroxiácido. Estudios Clínicos: Terapia combinada para el tratamiento de primera línea del carcinoma colorrectal metastásico: Irinotecan en combinación con cetuximab en pacientes sin tratamiento previo: EMR 62 202-013: Este estudio aleatorizado en pacientes con cáncer colorrectal metastásico que no recibieron tratamiento previo para la enfermedad metastásica comparó la combinación de cetuximab e irinotecan más 5-fluorouracilo en infusión / ácido folínico (5-FU/FA) (599 pacientes) con la misma quimioterapia sola (599 pacientes). La proporción de pacientes con tumores KRAS de tipo salvaje de la población de pacientes evaluables para el estatus KRAS comprendió el 64%. Los datos de eficacia generados en este estudio se resumen en la Tabla 1 a continuación: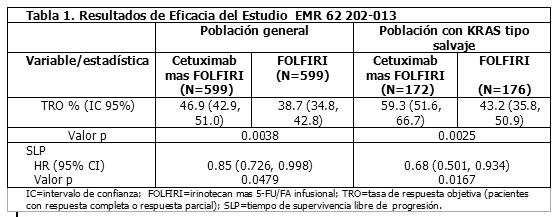
Irinotecan en combinación con bevacizumab: Un estudio clínico de fase III, aleatorizado, doble ciego, activo-controlado, evaluó al bevacizumab en combinación con irinotecan/5-FU/AF como tratamiento de primera-línea para el carcinoma metastático del colon o recto (Estudio AVF2107g). La adición de bevacizumab a la combinación de irinotecan/5-FU/AF resultó en un aumento estadísticamente significativo en la sobrevida total. El beneficio clínico, medido por la sobrevida total, fue observado en todos los subgrupos preespecificados de pacientes, incluyendo a los definidos por edad, sexo, estatus de desempeño, localización del tumor primario, número de órganos implicados y duración de la enfermedad metastásica. Refiérase también a la información para prescribir completa del bevacizumab. Los resultados de eficacia del Estudio AVF2107, están resumidos en la siguiente tabla 2: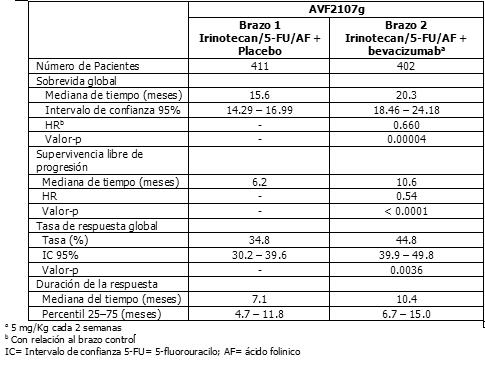
Irinotecan en combinación con capecitabina: Datos de un estudio aleatorizado controlado en fase III (CAIRO) sustentan el uso de capecitabina en una dosis inicial de 1,000 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan para el tratamiento de primera línea de pacientes con cáncer colorrectal metastásico. 820 pacientes fueron seleccionados aleatoriamente para recibir tratamiento secuencial (n=410) o tratamiento combinado (n=410). El tratamiento secuencial consistió en un tratamiento de primera línea con capecitabina (1250 mg/m2 dos veces diariamente por 14 días), el de segunda línea con irinotecan (350 mg/m2 el día 1), y el de tercera línea combinar capecitabina (1000 mg/m2 dos veces al día por 14 días) con oxaliplatin (130 mg/m2 el día 1). El tratamiento combinado consistió en tratamiento de primera línea de capecitabina (1000 mg/m2 dos veces diariamente por 14 días) combinado con irinotecan (250 mg/m2 el día 1) (XELIRI) y el de segunda línea capecitabina (1000 mg/m2 dos veces al día por 14 días) más oxaliplatino (130 mg/m2 el día 1). Todos los ciclos de tratamiento se registraron en intervalos de 3 semanas. En el tratamiento de primera línea la mediana de la supervivencia sin progresión en la población con intención de tratamiento fue 5.8 meses (95% CI, 5.1-6.2 meses) para la monoterapia de capecitabine y 7.8 meses (95% CI, 7.0-8.3 meses) para XELIRI (p=0.0002). Los datos de un análisis intermedio de un estudio multicéntrico, de selección aleatoria, controlado en fase II (AIO KRK 0604) sustentan el uso de capecitabina a una dosis de inicio de 800 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan y bevacizumab para el tratamiento de primera línea de pacientes con cáncer colorrectal metastásico. 115 pacientes fueron seleccionados aleatoriamente para el tratamiento con capecitabine combinado con irinotecan (XELIRI) y bevacizumab: capecitabina (800 mg/m2 dos veces al día por 2 semanas seguido de un periodo de descanso de 7 días), irinotecan (200 mg/m2 como infusión de 30 minutos el día 1 cada 3 semanas),y bevacizumab (7.5 mg/kg como una infusión de 30 a 90 minutos el día 1 cada 3 semanas); un total de 118 pacientes fueron seleccionados aleatoriamente para el tratamiento con capecitabine combinado con oxaliplatino más bevacizumab; capecitabina (1000 mg/m2 dos veces al día por 2 semanas seguido por un periodo de descanso de 7 días), oxaliplatino (130 mg/m2 como una infusión de 2 horas el día 1 cada 3 semanas) y bevacizumab (7.5 mg/kg como una infusión de 30 a 90 minutos en día 1 cada 3 semanas). La supervivencia libre de progresión a los 6 meses en la población con intención de tratamiento fue 80% (XELIRI más bevacizumab) versus 74% (XELOX más bevacizumab). El índice de respuesta global (respuesta completa más respuesta parcial) fue 45% (XELOX más bevacizumab) versus 47% (XELIRI más bevacizumab). Terapia combinada para el tratamiento de segunda línea del carcinoma colorrectal metastásico: Irinotecan en combinación con cetuximab después de la falla de la terapia citotóxica que incluye al irinotecan: La eficacia de la combinación de cetuximab con irinotecan fue investigada en dos estudios clínicos. Un total de 356 pacientes con cáncer colorrectal metastásico con expresión de EGFR que tuvieron recientemente falla en la terapia citotóxica que incluyera irinotecan y tenían un mínimo en el estatus de desempeño de Karnofsky de 60, pero la mayoría con un estado de desempeño de Karnofsky de ≥80 recibieron el tratamiento combinado. EMR 62 202-007: Este estudio aleatorizado comparó la combinación de cetuximab e irinotecan (218 pacientes) con monoterapia de cetuximab (111 pacientes). IMCL CP02-9923: Este estudio de ramificación abierta investigó la terapia de combinación en 138 pacientes. Los datos de eficacia de estos estudios se resumen en la Tabla 3 a continuación: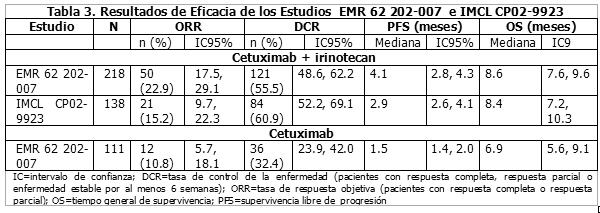
La eficacia de la combinación de cetuximab con irinotecan fue superior al de la monoterapia con cetuximab, en términos de tasa de respuesta objetiva (ORR, siglas en inglés), tasa de control de la enfermedad (DCR, siglás en inglés) y supervivencia sin progresión (PFS, siglas en inglés). En el estudio aleatorizado, no se demostraron efectos en la supervivencia general (tasa de riesgo 0.91, p=0.48).
Contraindicaciones: El irinotecan está contraindicado en pacientes con hipersensibilidad conocida a la droga o a sus excipientes. (Ver la sección Precauciones Generales - Reacciones de Hipersensibilidad).
Precauciones generales: Administración: El irinotecan solamente se debe administrar bajo la supervisión de un médico con experiencia en el uso de agentes quimioterapéuticos para el tratamiento del cáncer. El manejo adecuado de las complicaciones solo es posible, cuando se puede acceder fácilmente al diagnóstico adecuado e instalaciones para el tratamiento. El irinotecan se prescribirá solamente en los siguientes casos, después de haber sopesado los beneficios esperados contra los riesgos terapéuticos factibles: En pacientes que presenten un factor de riesgo, particularmente aquellos con un estado funcional de la OMS=2. En las pocas y raras circunstancias donde se considere improbable que los pacientes cumplan las recomendaciones relativas al manejo de los eventos adversos (necesidad de un tratamiento antidiarreico inmediato y prolongado, combinado con ingesta alta de líquidos al inicio de diarrea tardía). Para estos pacientes se recomienda una supervisión hospitalaria estricta. Síntomas colinérgicos. Los pacientes pueden tener síntomas colinérgicos de rinitis, salivación aumentada, miosis, lagrimeo, diaforesis, rubor (vasodilatación), bradicardia e hiperperistaltismo intestinal que cause calambres abdominales y diarrea temprana (es decir, la diarrea que ocurra generalmente durante o dentro de 8 horas de la administración del irinotecan). Estos síntomas se pueden observar durante o poco después de la infusión del irinotecan, y se cree están relacionados con la actividad anticolinesterasa del irinotecan inalterado y se espera que ocurran más frecuentemente con dosis más altas de irinotecan. Se debe considerar la administración terapéutica o profiláctica de 0,25 a 1 mg de atropina intravenosa o subcutánea (a menos que esté contraindicada), en los pacientes que experimenten síntomas colinérgicos. Extravasación. Aunque el irinotecan no es conocido como vesicante, se debe tener cuidado de evitar la extravasación y se debe vigilar el sitio de la infusión para detectar signos de inflamación. Si ocurre extravasación, se recomiendan lavado del sitio y aplicación local de hielo. Hepáticas. En estudios clínicos se observaron anormalidades de las enzimas hepáticas grado 3 ó 4, de los Criterios de Toxicidad Comunes (CTC) del Instituto Nacional de Cáncer (NCI, siglas en inglés), en menos del 10% de los pacientes. Estos eventos ocurren típicamente en los pacientes con metástasis hepáticas conocidas y no han sido relacionados claramente con el irinotecan. Hematológicas. Comúnmente el irinotecan causa neutropenia, leucopenia y anemia, cualquiera de las cuales pueden ser severa y por ello no se debe usar en pacientes con insuficiencia severa de la médula ósea. La trombocitopenia seria es poco común. En los estudios clínicos, la frecuencia de neutropenia grado 3 y 4 del NCI fue significativamente mayor en los pacientes que habían recibido previamente radioterapia pélvica/abdominal, que en los pacientes que no habían sido expuestos a esta terapia. Los pacientes con niveles basales de bilirrubina sérica total de 1,0 mg/dL o más, también tuvieron una probabilidad significativamente mayor de neutropenia grado 3 ó 4 en el primer ciclo de la terapia, que los pacientes con niveles de bilirrubina por debajo de 1,0 mg/dL. No hubo diferencias significativas entre la edad y el sexo en la frecuencia de neutropenia grado 3 ó 4. (Ver la sección Precauciones Generales - Poblaciones Especiales, Insuficiencia Hepática y la sección Dosis y Vía de Administración - Regímenes de Agente Único o de Combinación de Agentes). La fiebre neutropénica (neutropenia recurrente grado 4 del NCI y fiebre grado ≥2), ocurrió en menos del 10% de los pacientes en los estudios clínicos; sin embargo, en los pacientes tratados con irinotecan se han reportado muertes por septicemia después de mielosupresión severa. Las complicaciones neutropénicas se deben manejar con rapidez y con soporte antibiótico. Si ocurre fiebre neutropénica, o si el recuento absoluto de neutrófilos cae por debajo de 1.000/mm3, se debe suspender temporalmente la terapia con irinotecan. La dosis de irinotecan se debe disminuir, si ocurre neutropenia clínicamente significativa (Ver la sección Dosis y Vía de Administración, sección Recomendaciones para Modificar las Dosis). Pacientes con Actividad UGT1A1 Reducida. La conversión metabólica de irinotecan al metabolito activo SN-38 está mediada por las enzimas carboxilesterasa y ocurre principalmente en el hígado. El SN-38 experimenta subsecuentemente conjugación para formar el metabolito glucurónido inactivo SN-38G. Esta reacción de glucuronidación está mediada principalmente por uridin difosfato-glucuronosil transferasa 1A1 (UGT1A1), la cual está codificada por el gen UGT1A1. El gen UGT1A1 es altamente polimórfico, resultando en capacidades metabólicas variables entre individuos. Una variación específica del gen UGT1A1 incluye un polimorfismo en la región promotora conocida como el alelo variante UGT1A1 28. Esta variante y otras deficiencias congénitas en la expresión UGT1A1 (tales como el síndrome Crigler-Najjar y de Gilbert) están asociadas con la actividad enzimática reducida y la exposición sistémica incrementada a SN-38. Se observaron concentraciones superiores en el plasma de SN-38 en individuos homocigotos para el alelo UGT1A1*28 (también referido como genotipo 7/7 UGT1A1) versus pacientes que tienen uno o dos alelos de tipo salvaje. Los datos del meta-análisis de nueve estudios involucrando un total de 821 pacientes indican que los individuos con síndrome Crigler-Najjar (tipos 1 y 2) o los que son homocigotos para el alelo UGT1A1*28 (síndrome de Gilbert) tienen incremento en el riesgo de toxicidad hematológica (grados 3 y 4) después de la administración de irinotecan en dosis moderadas o altas ( > 150 mg/m2). No se estableció una relación entre el genotipo UGT1A1 y la ocurrencia de diarrea inducida por irinotecan. En los pacientes conocidos por ser homocigotos para UGT1A1*28 se les debe administrar la dosis de inicio de irinotecan indicada normalmente. Sin embargo, estos pacientes deben ser monitoreados por toxicidades hematológicas. Se debe considerar una dosis de inicio reducida de irinotecan en pacientes que hayan experimentado toxicidad hematológica con el tratamiento previo. La reducción exacta en la dosis de inicio en esta población de pacientes no ha sido establecida y cualquier modificación subsecuente de la dosis se debe basar en la tolerancia individual del paciente al tratamiento. Reacciones de hipersensibilidad. Se han reportado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas/anafilactoides severas. Efectos inmunodepresores / Aumento de la Susceptibilidad a Infecciones. La administración de vacunas vivas o vivas-atenuadas en pacientes inmunocomprometidos por agentes quimioterapéuticos, incluido el irinotecan, puede resultar en infecciones serias o fatales. La vacunación con una vacuna viva debe evitarse en pacientes que reciben irinotecan. Las vacunas muertas o inactivadas pueden administrarse; sin embargo, la respuesta a tales vacunas podría verse disminuida. Diarrea tardía. La diarrea tardía (que generalmente ocurre después de más de 8 horas de la administración del irinotecan) puede ser prolongada, causar deshidratación, desequilibrio electrolítico o septicemia y poner en peligro la vida. En los estudios clínicos que utilizaron el régimen de dosificación cada 3-semanas, la mediana del tiempo para el inicio de la diarrea tardía fue de 5 días, después de la infusión del irinotecan. En los estudios clínicos que evaluaron el régimen de dosificación semanal, la mediana del tiempo para el inicio de la diarrea tardía fue de 11 días, después de la administración del irinotecan. Para los pacientes que empezaron el tratamiento con una dosis semanal de 125 mg/m2, la mediana de la duración de cualquier grado de diarrea tardía fue de 3 días. Entre los pacientes tratados con la dosis semanal de 125 mg/m2 que experimentaron diarrea tardía grado 3 ó 4, la mediana de la duración de todo el episodio de diarrea fue 7 días. Los resultados de un estudio prospectivo del régimen de dosificación semanal no evidenciaron ninguna diferencia en la tasa de diarrea tardía en los pacientes con ≥56 años de edad, que los pacientes de < 65 años de edad. Sin embargo, los pacientes de más de 65 años deben ser monitoreados con cuidado por un mayor riesgo de diarrea inicial observada en esta población. Se ha observado ulceración del colon, algunas veces con sangrado, en asociación con la diarrea inducida por el irinotecan. La diarrea tardía se debe tratar inmediatamente con loperamida apenas ocurra el primer episodio de heces amorfas o sueltas, o en el momento de iniciarse movimientos intestinales más frecuentes que los normalmente esperados para el paciente. El régimen de dosificación recomendado para la loperamida, es de 4 mg en el momento de iniciarse la diarrea tardía y posteriormente 2 mg cada 2 horas, hasta que el paciente esté sin diarrea por lo menos 12 horas. Durante la noche, el paciente puede tomar 4 mg de loperamida cada 4 horas. No se recomienda el uso de loperamida por más de 48 horas consecutivas en esas dosis por el riesgo de íleo paralítico, pero tampoco por menos de 12 horas. No se recomienda la premedicación con loperamida. Los pacientes con diarrea severa deben ser monitoreados cuidadosamente y se les debe administrar reemplazo de líquidos y electrolitos si se deshidratan, así como soporte antibiótico si desarrollan íleo, fiebre o neutropenia severa. Además del tratamiento con antibióticos, se recomienda hospitalización para el control de la diarrea, en los siguientes casos: Diarrea asociada a fiebre, Diarrea severa (que requiere hidratación intravenosa), Pacientes con vómito asociado a la diarrea tardía. Diarrea que persista por más de 48 horas, después del inicio de una terapia con dosis altas de loperamida. Después del primer tratamiento, se deben retrasar los tratamientos semanales subsiguientes de quimioterapia de los pacientes, hasta que su función intestinal retorne a un nivel pretratamiento durante 24 horas por lo menos, sin necesidad de medicación antidiarreica. Si ocurre diarrea grado 2, 3 ó 4 del NCI, se deben disminuir las dosis subsiguientes del irinotecan dentro del ciclo en curso (Ver la sección Dosis y Vía de Administración, sección Recomendaciones para Modificar las Dosis). Enfermedad intestinal inflamatoria crónica y/u obstrucción intestinal. Los pacientes no se deben tratar con irinotecan, hasta la resolución de la obstrucción intestinal. Náusea y Vómito. El irinotecan es emetogénico. La náusea y el vómito pueden ser severos y usualmente ocurren durante o poco después de la infusión del irinotecan. Se recomienda que los pacientes reciban premedicación con agentes antieméticos. Los agentes antieméticos se deben administrar el mismo día del tratamiento, empezando por lo menos 30 minutos antes de la administración del irinotecan. Además, los médicos deben considerar prescribir a sus pacientes un régimen antiemético para uso subsiguiente, según se requiera. Los pacientes con vómitos asociados a la diarrea demorada (o sea, tardía), deben ser hospitalizados lo más pronto posible para tratamiento. Neurológicas. Se ha observado mareo, que algunas veces podría representar una evidencia sintomática de hipotensión ortostática en los pacientes con deshidratación. Renales. Se han observado aumentos en los niveles de creatinina sérica o del nitrógeno ureico en sangre. Hubo casos de insuficiencia renal aguda. Generalmente estos eventos se han atribuido a la deshidratación relacionada con la náusea, vómito o diarrea. También se han reportado casos raros de disfunción renal, debida al síndrome de lisis tumoral. Respiratorias. Se ha observado disnea grado 3 ó 4 del NCI. No se sabe en qué grado pudieron haber estado implicadas en la disnea, una malignidad pulmonar u otra enfermedad pulmonar preexistente. En los primeros estudios realizados en Japón, se observó en un pequeño porcentaje de pacientes un síndrome pulmonar que potencialmente pone en peligro la vida, consistente en disnea, fiebre y un patrón reticulonodular en las radiografías de tórax. Fue difícil evaluar la contribución del irinotecan en estos eventos preliminares, porque los pacientes también presentaban tumores pulmonares y algunos de ellos tenían una enfermedad pulmonar no-maligna preexistente. La enfermedad pulmonar intersticial con manifestación de infiltrados pulmonares es poco común durante el tratamiento con irinotecan. La enfermedad pulmonar intersticial puede ser fatal. Los factores de riesgo asociados con el desarrollo de enfermedad pulmonar intersticial incluyen una enfermedad pulmonar preexistente, el uso de drogas neumotóxicas, la radioterapia y los factores estimulantes de colonias. Los pacientes con factores de riesgo, deben ser monitoreados de cerca para identificar síntomas respiratorios, antes y después del tratamiento con irinotecan. Otros. Como este producto contiene sorbitol, es inadecuado en la intolerancia a la fructosa hereditaria. Poblaciones Especiales: Pediátrica. La eficacia del irinotecan en pacientes pediátricos no se ha establecido. (Ver la sección Farmacocinética y Farmacodinamia - Farmacocinética en Poblaciones Especiales, Pediátrica). Se evaluaron los resultados de dos estudios abiertos, de un solo brazo de tratamiento. Ciento setenta niños con tumores sólidos refractarios se reclutaron en un estudio de fase 2, donde se infundieron 50 mg/m2 de irinotecan durante 5 días consecutivos, cada 3 semanas. La neutropenia grado 3-4 se presentó en 54 de los pacientes (31,8%). En 15 pacientes, la neutropenia se vio complicada con fiebre (8,8%). En 35 (20,6%) de los pacientes, se observó diarrea grado 3-4. Este perfil de eventos adversos, es comparable al observado en adultos. En el segundo estudio de fase 2, a 21 niños con rabdomiosarcoma no tratado anteriormente, se les infundieron 20 mg/m2 de irinotecan por 5 días consecutivos, en las semanas 0, 1, 3 y 4. Esta terapia de agente único, se continuó con una terapia multimodal. El reclutamiento en la fase de irinotecan como agente único se interrumpió, debido a la alta tasa de enfermedad progresiva (28,6%) y de muertes tempranas (14%). En este estudio, el perfil de eventos adversos fue diferente al observado en adultos; los eventos adversos más significativos de grado 3 o 4 fueron deshidratación experimentada por 6 pacientes (28,6%), asociada con hipopotasemia severa en 5 pacientes (23,8%) e hiponatremia en 3 pacientes (14,3%); adicionalmente, en 5 pacientes (23,8%) se reportó infección de grado 3-4 (entre todos los cursos de terapia e independientemente de la relación causativa). Geriátrica. Para esta población se pueden aplicar recomendaciones de dosificación específicas, dependiendo del régimen utilizado (Ver la sección Dosis y Vía de Administración). Insuficiencia Hepática. En pacientes con hiperbilirrubinemia, la depuración del irinotecan está disminuida (Ver la sección Farmacocinética y Farmacodinamia - Farmacocinética en Poblaciones Especiales) y, en consecuencia, aumenta el riesgo de hematotoxicidad. El uso del irinotecan en pacientes con una concentración de bilirrubina sérica total > 3.0 x límite superior normal institucional (LSNI), administrado como agente único con el régimen de dosificación una vez-cada-3-semanas, no se ha establecido (Ver la sección Dosis y Vía de Administración - Poblaciones Especiales). La función hepática debe ser monitoreada antes del inicio del tratamiento y mensualmente, o como esté indicado clínicamente. Radioterapia. Los pacientes que han recibido previamente irradiación pélvica/abdominal, tienen un mayor riesgo de mielosupresión después de la administración de irinotecan. Los médicos deben ser precavidos al tratar a pacientes que hayan tenido irradiación extensa previa. Para esta población se pueden aplicar recomendaciones de dosificación específicas, dependiendo del régimen utilizado (Ver la sección Dosis y Vía de Administración). Estatus de desempeño. Los pacientes con un status de desempeño pobre, tienen un mayor riesgo de eventos adversos relacionados con el irinotecan. Se pueden aplicar recomendaciones de dosificación específicas, para los pacientes con un estado de condición física de 2 del Grupo Cooperativo Oncológico del Este (ECOG, siglas en inglés), dependiendo del régimen utilizado (Ver la sección Dosis y Vía de Administración). Los pacientes con status de desempeño de 3 ó 4 no deben recibir irinotecan. Entre los pacientes que recibieron irinotecan/5-FU/LV ó 5-FU/LV en los estudios clínicos que compararon a esos agentes, se observaron tasas más altas de hospitalización, fiebre neutropénica, tromboembolismo, discontinuación en el primer ciclo de tratamiento y muertes tempranas en los que tuvieron un estado funcional de 2, que en los que tuvieron un status de desempeño de 0 ó 1, en el estado basal. Cáncer gástrico. Los pacientes con cáncer gástrico experimentan aparentemente mayor mielosupresión y otras toxicidades, cuando son tratados con irinotecan. En estos pacientes se debe considerar una dosis inicial menor (Ver la sección Dosis y Vía de Administración), Efectos sobre la capacidad para manejar y utilizar máquinas: El efecto del irinotecan sobre la capacidad para manejar o usar maquinaria no ha sido evaluado. Sin embargo, los pacientes deben ser advertidos sobre la posibilidad de que se mareen o tengan alteraciones visuales, que podría ocurrir dentro de las 24 horas de la administración del irinotecan, recomendándoles no manejar ni operar maquinarias, si se presentan esos síntomas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: El irinotecan es teratogénico en ratas y conejos (Ver la sección Precauciones en Relación con Efecto de Carcinogénesis, Mutagénesis y Teratogénesis sobre Fertilidad). El irinotecan puede causar daño fetal, si es administrado a una mujer embarazada. No se han realizado estudios adecuados y bien controlados con irinotecan en mujeres embarazadas. Si el fármaco se usa durante el embarazo, o si la paciente queda embarazada mientras esté recibiendo este fármaco, ella será advertida del peligro potencial para el feto. A las mujeres en capacidad de concebir se les debe advertir que deben evitar quedar embarazadas, mientras estén recibiendo tratamiento con irinotecan. Lactancia: En ratas, la radioactividad apareció en la leche a los 5 minutos después de la administración intravenosa de irinotecan marcado radiactivamente y se concentró hasta 65 veces, con relación a las concentraciones plasmáticas, a las 4 horas después de la administración. Como muchos fármacos se excretan en la leche humana y considerando el potencial de reacciones adversas serias en los infantes lactantes, se recomienda descontinuar la lactancia cuando se esté recibiendo terapia con irinotecan.
Reacciones secundarias y adversas: Estudios Clínicos: Los datos de eventos adversos del programa de estudios clínicos en cáncer colorrectal metastásico que recurre o progresa después de terapia a base de 5-FU (segunda línea), fue recolectada y analizada exhaustivamente y se presenta a continuación (la población de pacientes se describe más adelante). Se espera que las reacciones adversas para otras indicaciones sean similares a los observados para la terapia de segunda línea del cáncer colorrectal. Las reacciones adversas detalladas en esta sección se refieren al irinotecan. No existe ninguna prueba de que el perfil de seguridad del irinotecan se vea influenciado por el cetuximab o viceversa. En combinación con el cetuximab, las reacciones adversas adicionales reportadas fueron las esperadas con el cetuximab (tales como erupción acneiforme). Por lo tanto, refiérase también a la información de prescripción completa del cetuximab. La hipertensión grado 3 fue el principal riesgo significativo implicado por la adición de bevacizumab a bolo de irinotecan/5-FU/AF. Además, con este régimen hubo un pequeño aumento en los eventos adversos diarrea y leucopenia de Grado 3/4 de la quimioterapia, en comparación con los pacientes que recibían solamente bolo de irinotecan/5-FU/AF. Para otra información sobre reacciones adversas en combinación con el bevacizumab, refiérase a la información de prescripción completa del bevacizumab. Las reacciones adversas de la droga reportadas en pacientes tratados con la combinación de irinotecan más capecitabine, adicional a los vistos con monoterapia de capecitabine u observados a una frecuencia grupal superior con la combinación comparados con la monoterapia de capecitabine incluye: Muy común, todos los grados: trombosis/embolismo. Común, todos los grados: reacción de hipersensibilidad, isquemia cardiaca / infarto. Común, grado 3 y grado 4: neutropenia febril. Para información más completa sobre las reacciones adversas del capecitabine, refiérase a la información de prescripción de capecitabine. Las reacciones adversas del fármaco de Grado 3 y Grado 4 reportadas en pacientes tratados con irinotecan en combinación con capecitabine y bevacizumab, adicionalmente a lo observado con la monoterapia de capecitabine o visto a una frecuencia grupal superior con la combinación comparado con la monoterapia de ca
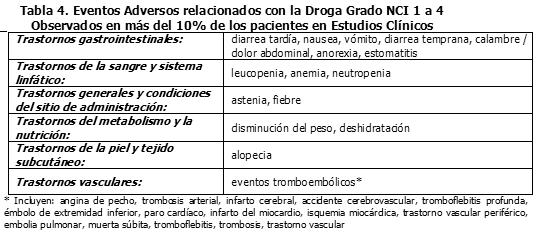
Los eventos adversos grado 3 ó 4 del NCI reportados en los estudios clínicos de los regímenes de dosificación semanal y una vez cada 3 semanas (N= 620), están listados en las Tablas 4 a la 5.

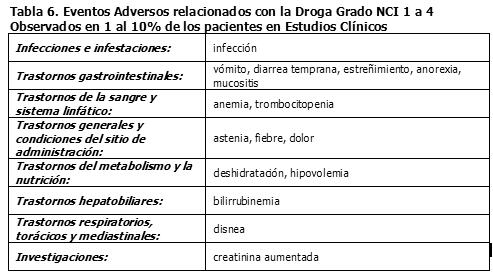
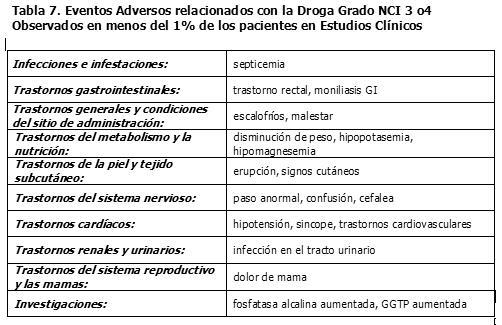
Los siguientes eventos adicionales relacionados con el fármaco, fueron reportados en estudios clínicos con irinotecan, pero no alcanzaron los criterios definidos anteriormente al evento relacionado con el fármaco de grado NCI 1-4 en > 10% de los pacientes o de los eventos relacionados con el fármaco de grado NCI 3 ó 4: rinitis, salivación aumentada, miosis, lagrimeo, diaforesis, rubefacción, bradicardia, mareo, extravasación, síndrome de lisis tumoral y ulceración del colon. Vigilancia Post-Comercialización: Trastornos cardíacos: Se han observado eventos isquémicos miocárdicos después del tratamiento con irinotecan, predominantemente en pacientes con enfermedad cardíaca subyacente, otros factores de riesgo conocidos de enfermedad cardíaca o quimioterapia citotóxica previa. (Ver también Tabla 4, eventos tromboembólicos). Trastornos gastrointestinales: Se reportaron casos infrecuentes de obstrucción intestinal, íleo o hemorragia gastrointestinal y casos raros de colitis, incluyendo tiflitis, colitis isquémica y colitis ulcerativa. En algunos casos, la colitis se vio complicada por ulceración, sangrado, íleo o infección. También se han reportado casos de íleo sin colitis precedente. Se reportaron casos raros de perforación intestinal. Se han observado casos raros de pancreatitis sintomática o de enzimas pancreáticas elevadas asintomáticas. Hipovolemia: Han ocurrido casos raros de insuficiencia renal y deterioro renal agudo, generalmente en pacientes que se infectaron y/o tuvieron depleción de volumen por toxicidades gastrointestinales severas. Se han observado casos infrecuentes de insuficiencia renal, hipotensión o insuficiencia circulatoria, en pacientes que experimentaron episodios de deshidratación asociada con diarrea y/o vómito o de septicemia. Trastornos del sistema inmunológico: Se han reportado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas o anafilactoides severas (Ver la sección Precauciones Generales). Trastornos musculoesqueléticos y del tejido conectivo: Se han reportado efectos tempranos, tales como contracciones o calambres musculares y parestesia. Trastornos del sistema nervioso: Se han reportado trastornos del habla, generalmente transitorios en pacientes tratados con irinotecan; en algunos casos, el evento fue atribuido al síndrome colinérgico observado durante o inmediatamente después de la infusión de irinotecan. Trastornos respiratorios, torácicos y mediastinales: La enfermedad pulmonar intersticial que se presenta como infiltrados pulmonares no es común durante la terapia con irinotecan. Se han reportado efectos tempranos, tales como disnea (Ver la sección Precauciones Generales). También se han reportado hipos. Investigaciones: Se han reportado casos raros de hiponatremia, principalmente relacionados con diarrea y vómito. Raras veces se han reportado aumentos transitorios y de leves a moderados en los niveles de transaminasas (o sea, AST y ALT), en ausencia de metástasis hepática progresiva.
Interacciones medicamentosas y de otro género: Agentes Bloqueadores Neuromusculares: No se puede excluir interacción entre el irinotecan y agentes bloqueadores neuromusculares, ya que el irinotecan tiene actividad anticolinesterasa. Los fármacos con actividad anticolinesterasa pueden prolongar los efectos de bloqueo neuromuscular del suxametonio y puede verse antagonizado el bloqueo neuromuscular de las drogas no-despolarizantes. Agentes Antineoplásicos: Cabe esperar que los efectos adversos del irinotecan, tales como mielosupresión y diarrea, sean exacerbados por otros agentes antineoplásicos que tengan un perfil de efectos adversos similar. Dexametasona: Se ha reportado linfocitopenia en pacientes recibiendo irinotecan y es posible que la administración de dexametasona como profilaxis antiemética, pueda aumentar la probabilidad de linfocitopenia. Sin embargo, no se han observado infecciones oportunistas serias y ninguna complicación ha sido atribuida específicamente a linfocitopenia. Se ha observado hiperglucemia o evidencia de intolerancia a la glucosa en pacientes con antecedentes de diabetes mellitus, antes de la administración del irinotecan. Es probable que la dexametasona, administrada como profilaxis antiemética, haya contribuido a la hiperglucemia en algunos pacientes. Laxantes: Se prevé que el uso de laxantes durante la terapia con irinotecan empeore la incidencia o severidad de la diarrea. Diuréticos: El irinotecan puede inducir deshidratación secundaria al vómito y/o la diarrea. Sería deseable que el médico suspenda los diuréticos durante el tratamiento con irinotecan y durante los periodos de vómito y diarrea activos. Anticonvulsivantes: La administración concomitante de drogas anticonvulsivantes que inducen la CYP3A (por ej., carbamazepina, fenobarbital o fenitoína), resulta en una exposición reducida al metabolito activo SN-38. En los pacientes que requieran tratamiento anticonvulsivante, se debe considerar el inicio o la sustitución por anticonvulsivantes no inductores, por lo menos una semana antes del inicio del tratamiento con irinotecan. Ketoconazol: La depuración del irinotecan se ve muy reducida en los pacientes que reciben concomitantemente ketoconazol, lo que resulta en un aumento en la exposición al SN-38. El ketoconazol debe ser discontinuado por lo menos 1 semana antes de iniciar el tratamiento con irinotecan y no se debe administrar durante el tratamiento con el mismo. Hierba de San Juan (St. John's Wort, Hypericum perforatum): La exposición al metabolito activo SN-38 está reducida en pacientes que reciben concomitantemente la Hierba de San Juan. Esta debe ser discontinuada por lo menos 1 semana antes del primer ciclo de irinotecan y no se debe administrar durante la terapia con irinotecan. Sulfato de Atazanavir: La administración concomitante del sulfato de atazanavir, un inhibidor de la CYP3A4 y UGT1A1, tiene el potencial de aumentar la exposición sistémica al SN-38, el metabolito activo del irinotecan. Los médicos deben tomar en cuenta esto, cuando coadministren estas drogas. Bevacizumab: En un estudio, las concentraciones plasmáticas del irinotecan eran similares en los pacientes que recibían irinotecan/5-FU/AF solo y en combinación con bevacizumab. Las concentraciones de SN-38, el metabolito activo del irinotecan, fueron analizadas en un subgrupo de pacientes (aproximadamente 30 por brazo de tratamiento). Las concentraciones de SN-38 estaban, en promedio, 33% más altas en los pacientes que recibían irinotecan/5-FU/AF en combinación con bevacizumab, que en los que recibían irinotecan/5-FU-AF solamente. Debido a la alta variabilidad inter-paciente y al muestreo limitado, no hay certeza de que el aumento observado en los niveles de SN-38 se haya debido al bevacizumab. Hubo un pequeño aumento en los eventos adversos de diarrea y leucopenia. Se reportaron más reducciones de la dosis del irinotecan entre los pacientes que recibían irinotecan/5-FU/AF en combinación con bevacizumab. A los pacientes que desarrollen diarrea severa, leucopenia, o neutropenia con bevacizumab e irinotecan en combinación, se les deben implementar las modificaciones de las dosis de irinotecan contempladas en la sección Dosis y Vía de Administración.
Alteraciones en los resultados de pruebas de laboratorio: Es recomendable un minucioso monitoreo de la cuenta de las células blancas de la sangre con diferencial, de hemoglobina y de la cuenta plaquetaria, antes de cada dosis de CAMPTOSAR®.
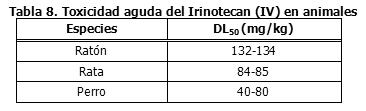
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Toxicología: La toxicidad intravenosa aguda del irinotecan en animales se muestra en la Tabla 8. Se observó letalidad después de dosis intravenosas únicas de irinotecan, de aproximadamente 111 mg/kg en ratones y 73 mg/kg en ratas (aproximadamente 2,6 y 3,4 veces la dosis humana recomendada de 125 mg/m2, respectivamente). La muerte se vio precedida por cianosis, temblores, distrés respiratorio y convulsiones. Los estudios de toxicidad subaguda mostraron que el irinotecan afecta los tejidos de proliferación celular rápida (médula ósea, epitelio intestinal, timo, bazo, ganglios linfáticos y testículos).
Carcinogenicidad / Mutagenicidad: No se han efectuado estudios de carcinogenicidad a largo plazo con irinotecan. Sin embargo, se administraron dosis intravenosas de 2 mg/kg o 25 mg/kg de irinotecan una vez por semana, durante 13 semanas en ratas (en estudios separados, la dosis de 25 mg/kg produjo una Cmáx y un AUC de irinotecan que fueron alrededor de 7,0 veces y 1,3 veces, respectivamente, los valores observados en pacientes que recibieron 125 mg/m2), dejándolas luego en recuperación durante 91 semanas. Bajo esas condiciones, hubo una tendencia lineal con la dosis, significativa para la incidencia de pólipos endometriales o uterinos y sarcomas estromales endometriales. Ni el irinotecan, ni el SN-38 fueron mutagénicos en el ensayo in vitro de Ames. Sin embargo, en el ensayo in vitro de aberración cromosómica de células de hámsteres Chinos, el irinotecan produjo un incremento significativo en la incidencia de aberraciones cromosómicas, en forma dependiente de la concentración. Adicionalmente, en el ensayo in vivo de micronúcleo de ratón, una sola dosis intraperitoneal de irinotecan en el rango de dosis de 2,5 a 200 mg/kg, causó un incremento significativo y dependiente de la dosis en la formación de eritrocitos policromáticos micronucleados y una disminución en la proporción reticulocitos/eritrocitos en las células de la médula ósea. Reproducción: No se observaron efectos adversos significativos sobre la fertilidad y el rendimiento reproductivo general, después de la administración intravenosa de irinotecan en dosis de hasta 6,0 mg/kg/día en ratas. Sin embargo, después de la administración de dosis múltiples diarias de irinotecan, se evidenció atrofia de los órganos reproductores masculinos, tanto en roedores con 20 mg/kg (que en estudios separados produjo una Cmáx y el ABC para el irinotecan de alrededor de 5 veces y 1 vez, respectivamente, los valores correspondientes observados en pacientes que recibieron 125 mg/m2), como en perros con 0,4 mg/kg (que en estudios separados produjo una Cmáx y un AUC para el irinotecan de alrededor de la mitad y 1/15°, respectivamente, los valores correspondientes observados en pacientes que recibieron 125 mg/m2). La radiactividad relacionada con el 14C-irinotecan atravesó la placenta de ratas, después de la administración intravenosa de 10 mg/kg (que en estudios separados produjo una Cmáx y el ABC el irinotecan de alrededor de 3 y 0,5 veces, respectivamente, de los valores correspondientes a pacientes recibiendo 125 mg/m2). El irinotecan fue teratogénico en ratas, en dosis superiores a 1,2 mg/kg/día (que en estudios separados produjo una Cmáx y el ABC para el irinotecan de alrededor de 2/3 y 1/40°, respectivamente, de los valores correspondientes a pacientes recibiendo 125 mg/m2) y en conejos en 6 mg/kg/día (alrededor de la mitad de la dosis semanal recomendada para humanos expresada en mg/m2). Los efectos teratogénicos incluyeron varios tipos de anomalías externas, viscerales y esqueléticas. El irinotecan administrado a ratas madres durante el período subsiguiente a la organogénesis, a través del destete en dosis de 6 mg/kg/día, causó una disminución en la capacidad de aprendizaje y disminución de los pesos corporales en los descendientes.
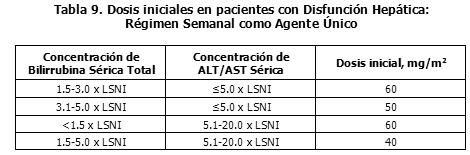
Dosis y vía de administración: Todas las dosis de irinotecan se deben administrar como una infusión intravenosa, durante 30 a 90 minutos. Regímenes de Dosificación como Agente Único: Los regímenes de dosis única se han estudiado extensamente para el cáncer colorrectal metastático. Estos regímenes se pueden usar en el tratamiento de pacientes con otros cánceres indicados (Ver Indicaciones Terapéuticas). Dosis Inicial: Régimen de Dosificación Semanal. La dosis inicial recomendada de irinotecan como agente único, es 125 mg/m2. Se puede considerar una dosis inicial menor (por ej., 100 mg/m2), para pacientes con alguna de las siguientes condiciones: radioterapia extensa previa, estatus de desempeño de 2, niveles de bilirrubina aumentados o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6-semanas, comprendiendo un tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas. Régimen de Dosificación una vez cada 2 semanas. La dosis inicial usualmente recomendada de irinotecan es 250 mg/m2 cada 2 semanas, por infusión intravenosa. Se puede considerar una dosis inicial menor (por ej., 200 mg/m2), para pacientes con alguna de las siguientes condiciones: 65 años o más de edad, radioterapia extensa previa, status de desempeño de 2, niveles de bilirrubina aumentados o cáncer gástrico. Régimen de Dosificación una vez cada 3 semanas. La dosis inicial usualmente recomendada de irinotecan para el régimen de dosificación una vez cada 3 semanas, es 350 mg/m2. Se puede considerar una dosis inicial menor (por ej., 300 mg/m2), para pacientes con alguna de las siguientes condiciones: 65 años o más de edad, radioterapia extensa previa, status de desempeño de 2, niveles de bilirrubina aumentados o cáncer gástrico. Poblaciones Especiales: Pacientes con la Función Hepática Deteriorada: En los pacientes con disfunción hepática, se recomiendan las siguientes dosis iniciales:

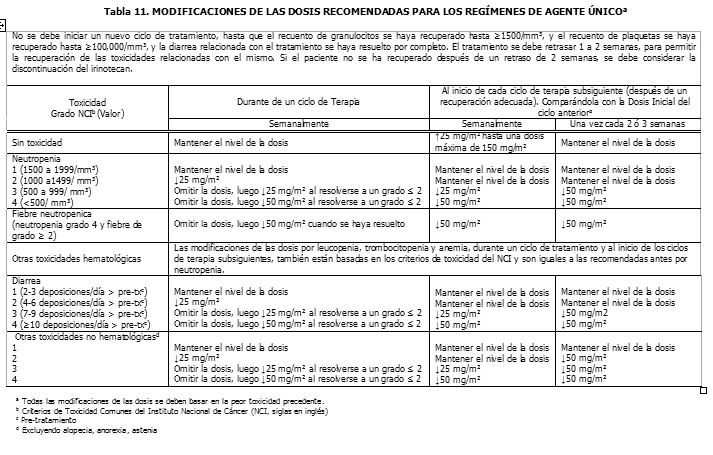
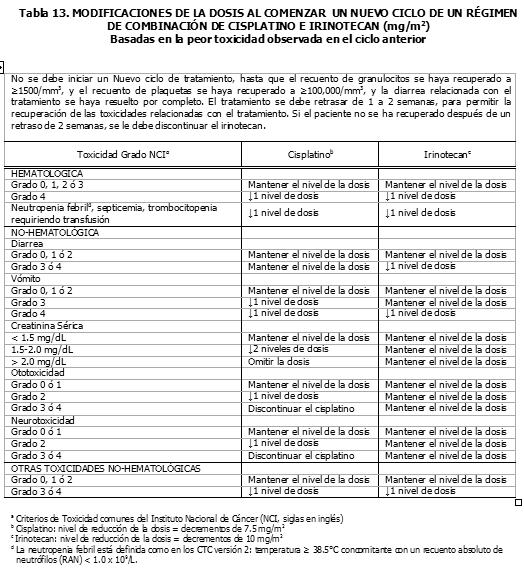
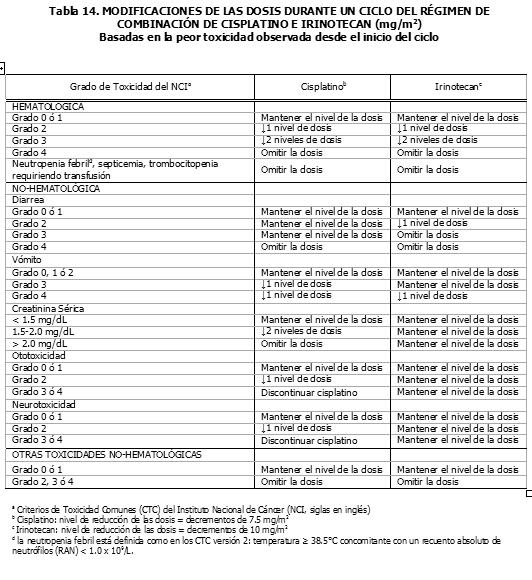
Pacientes con la Función Renal Deteriorada: No se han efectuado estudios en esta población (Ver la sección Farmacocinética y Farmacodinamia - Propiedades Farmacocinéticas: Farmacocinética en Poblaciones Especiales). Por lo tanto, se debe tomar precauciones en pacientes con la función renal deteriorada. No se recomienda el uso del irinotecan en pacientes en diálisis. Regímenes de Dosificación como Agente de Combinación: Dosis Inicial: Irinotecan en Combinación con 5-Fluorouracilo (5-FU) y Leucovorin en el Cronograma de cada 2 semanas. El uso del irinotecan en combinación con 5-FU y leucovorin, se recomienda en pacientes con cáncer colorrectal metastático. Para todos los regímenes, la dosis de leucovorin se debe administrar inmediatamente después del irinotecan, administrando 5-FU inmediatamente después de la recepción de leucovorin. Los regímenes actualmente recomendados se muestran a continuación: Régimen 1 (ciclo de 6 semanas con bolo de 5-FU/LV): Las dosis iniciales recomendadas son 125 mg/m2 de irinotecan, 500 mg/m2 de bolo 5-FU y 20 mg/m2 de bolo de leucovorin. Régimen 2 (ciclo de 6 semanas de 5-FU/LV en infusión): Las dosis iniciales recomendadas son 180 mg/m2 de irinotecan, 400 mg/m2 de bolo 5-FU, 600mg/m2 de 5FU en infusión y 200 mg/m2 de leucovorin. Se pueden considerar dosis iniciales menores de irinotecan (por ej., 100 mg/m2) y 5-FU (por ej., 400 mg/m2) para pacientes con alguna de las siguientes condiciones: 65 años o más de edad, radioterapia extensa previa, status de desempeño de 2, niveles de bilirrubina aumentados o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6-semanas, comprendiendo tratamiento durante 4-semanas, seguidos por un descanso de 2-semanas. Irinotecan en Combinación con Cisplatino. El irinotecan ha sido estudiado en combinación con cisplatino, para el cáncer pulmonar de célula no-pequeña y de célula pequeña, cáncer cervical, cáncer gástrico y cáncer esofágico. Este régimen se puede usar en pacientes con otros cánceres indicados, excepto para el cáncer colorrectal (Ver la sección Indicaciones Terapéuticas). La dosis inicial recomendada es 65 mg/m2 de irinotecan y 30 mg/m2 de cisplatino. Se puede considerar una dosis menor de irinotecan (ej., 50 mg/m2) para pacientes con alguna de las siguientes condiciones: 65 años o más de edad, radioterapia extensa previa, status de desempeño de 2, niveles de bilirrubina aumentados o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, comprendiendo tratamiento durante 4 semanas, seguidos por un descanso de 2 semanas. Irinotecan en Combinación con Cetuximab. Para la posología y método de administración concomitante con cetuximab, refiérase a la información de prescripción completa del cetuximab. Normalmente se usa la misma dosis de irinotecan, administrándola en los últimos ciclos del régimen que contiene irinotecan. El irinotecan no debe ser administrado antes de 1 hora después que haya finalizado la infusión de cetuximab. Irinotecan en Combinación con Bevacizumab. Para la posología y método de administración del bevacizumab, refiérase a la información de prescripción completa del bevacizumab. El bevacizumab se recomienda en combinación con el irinotecan (125 mg/m2) / bolo de 5-FU (500 mg/m2) / ácido folínico (20 mg/m2), administrado una vez a la semana durante 4 semanas, cada 6 semanas. Irinotecan en Combinación con Capecitabine. Para la posología y método de administración de capecitabine, ver sección Propiedades Farmacocinéticas y Farmacodinámicas - Propiedades farmacodinámicas y refiérase a la información de prescripción completa para capecitabine. El capecitabine está recomendado en combinación con irinotecan en una dosis inicial de 1000 mg/m2 por 2 semanas cada 3 semanas. Duración del Tratamiento: Para ambos regímenes, de agente único y de combinación de agentes, el tratamiento con ciclos adicionales de irinotecan se puede continuar indefinidamente en los pacientes que logren una respuesta tumoral o en aquellos cuyo cáncer permanezca estable. Los pacientes se deben monitorear a fondo para determinar toxicidad y se les debe suspender la terapia si se produce una toxicidad inaceptable, que no responda a la modificación de la dosis y a la atención médica de soporte rutinaria. Recomendaciones para las Modificaciones de las Dosis: Las modificaciones de las dosis recomendadas durante un ciclo de terapia y al inicio de cada ciclo de terapia subsiguiente, para los regímenes de agente único, están descritas en la Tabla 11. Estas recomendaciones están basadas en las toxicidades observadas más comúnmente con la administración del irinotecan. Para las modificaciones al inicio de un ciclo de terapia subsiguiente, la dosis del irinotecan se debe disminuir con relación a la dosis inicial del ciclo anterior. Las modificaciones de las dosis recomendadas durante un ciclo de terapia y al inicio de cada ciclo de terapia subsiguiente, para el irinotecan, 5-FU y leucovorin, están descritas en la Tabla 12. Las modificaciones de las dosis recomendadas del irinotecan y el cisplatino al inicio de cada ciclo de terapia están descritas en la Tabla 13, mientras que las modificaciones recomendadas para las dosis durante un ciclo de terapia, están descritas en la Tabla 14. Las recomendaciones para las modificaciones de las dosis de cetuximab cuando se administra en combinación con el irinotecan, deben seguirse de acuerdo con la información de prescripción completa del cetuximab. Refiérase a la información de prescripción completa del bevacizumab, cuando se administre en combinación con irinotecan/5-FU/AF. En combinación con capecitabine para pacientes con 65 años o más, se recomienda una reducción de la dosis inicial de capecitabine a 800 mg/m2 dos veces diariamente según la información de prescripción completa para capecitabine. Refiérase también a las recomendaciones para las modificaciones de dosis en el régimen de combinación dado en la información de prescripción completa para capecitabine. Todas las modificaciones de las dosis se deben basar en la peor toxicidad precedente. No se debe comenzar un nuevo ciclo de terapia, hasta que la toxicidad se haya recuperado hasta un grado 2 o menor. El tratamiento se puede retrasar 1 a 2 semanas, para permitir la recuperación de la toxicidad relacionada con el tratamiento. Si el paciente no se ha recuperado, se debe considerar la discontinuación del irinotecan.

Manifestaciones y manejo de la sobredosificación o ingesta accidental: Se han administrado dosis únicas de hasta 750 mg/m2 de irinotecan en pacientes con diversos cánceres. Los eventos adversos en estos pacientes fueron similares a los reportados con las dosis y regímenes recomendados. Se han producido reportes de sobredosificación en dosis de hasta aproximadamente el doble de la dosis terapéutica recomendada, que pudieron ser fatales. Las reacciones adversas más significativas reportadas, fueron neutropenia severa y diarrea severa. Se debe instituir el cuidado de soporte máximo, para prevenir la deshidratación debida a diarrea y tratar cualquier complicación infecciosa. No existe un antídoto conocido para la sobredosificación de irinotecan.
Presentación(es): Caja de cartón con un frasco vial de vidrio con 40 mg/2 mL. Caja de cartón con un frasco vial de vidrio con 100 mg/5 mL. Caja de cartón con un frasco vial de plástico ámbar con 40 mg/2 ml. Caja de cartón un frasco vial de plástico ámbar con 100 mg/5 ml. Caja de cartón un frasco vial de plástico ámbar con 300 mg/15 ml.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C. Las soluciones diluidas en dextrosa al 5% se deben conservar en refrigeración entre 2-8°C durante 48 horas. No se congele. Protéjase de la luz. La refrigeración de las soluciones diluidas con cloruro de sodio al 0.9% no es recomendable debido a una baja y esporádica incidencia de partículas visibles.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Este medicamento deberá ser administrado únicamente por médicos especialistas en Oncología y con experiencia en Quimioterapia Antineoplásica. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre durante el embarazo y la lactancia. Si no se administra todo el producto deséchese el sobrante. Léase el instructivo anexo.
Nombre y domicilio del laboratorio: Pfizer, S.A. de C.V. Km 63 Carretera México-Toluca, Zona Industrial, C.P. 50140, Toluca, México. México. ®Marca Registrada.
Número de registro del medicamento: 431M97 SSA IV
Clave de IPPA: 103300415D0179
Principios Activos de Camptosar
Patologías de Camptosar
Laboratorio que produce Camptosar
С 1win регистрация открывает доступ к миру азартных игр. Онлайн-казино гарантирует незабываемые эмоции и крупные выигрыши. Играйте в любимые азартные игры и выигрывайте с помощью pin up. Ваше время настало! Быстрое и рабочее мостбет зеркало обеспечивает надежный доступ к платформе в любое время и без проблем.


 Cambiar de país
Cambiar de país