VFEND®
PFIZER
Solución inyectable
Denominación genérica: Voriconazol.
Forma farmacéutica y formulación: Solución Inyectable. El frasco ámpula con liofilizado contiene: Voriconazol 200 mg. Excipiente cs.
Indicaciones terapéuticas: el voriconazol es un agente antimicótico triazólico de amplio espectro que tiene las siguientes indicaciones: tratamiento de aspergilosis invasiva. Tratamiento de candidemia en pacientes no neutropénicos. Tratamiento de infecciones invasivas graves por Candida (incluida la C. krusei). Tratamiento de candidiasis esofágica. Tratamiento de infecciones micóticas graves causadas por Scedosporium spp. y Fusarium spp. Tratamiento de otras infecciones micóticas graves en pacientes que tienen intolerancia o son refractarios a otras terapias. Prevención de brotes de infecciones micóticas en pacientes febriles de alto riesgo (pacientes con transplante alogénico de médula ósea, pacientes con recaída de leucemia).
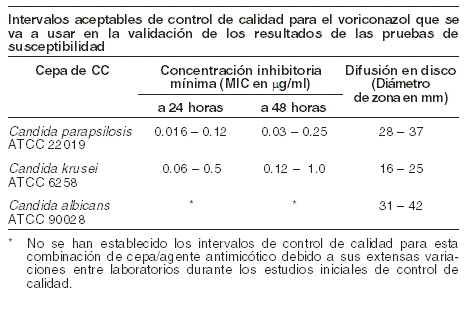
Farmacocinética y farmacodinamia: Propiedades farmacodinámicas: mecanismo de acción: in vitro, el voriconazol despliega actividad antimicótica de amplio espectro con potencia antimicótica contra especies de Candida (incluidos C. krusei resistente al fluconazol y cepas de C. glabrata y C. albicans resistentes) y actividad fungicida contra todas las especies examinadas de Aspergillus. Además, el voriconazol muestra actividad fungicida in vitro contra patógenos micóticos emergentes, incluidos aquellos como Scedosporium o Fusarium que tienen susceptibilidad limitada a los agentes antimicóticos existentes. Su modo de acción es la inhibición de la desmetilación del 14a-esterol mediada por el citocromo P450 del hongo, un paso esencial en la biosíntesis del ergosterol. En estudios con animales existe una correlación entre los valores de concentración inhibitoria mínima y eficacia contra micosis experimentales. Además, parece haber una correlación entre los valores de concentración inhibitoria mínima y el desenlace clínico para las especies de Candida. Microbiología: se demostró eficacia clínica para Aspergillus spp. incluidos A. flavus, A. fumigatus, A. terreus, A. niger, A. nidulans; Candida spp., incluidos C. albicans, C. glabrata, C. krusei, C. parapsilosis y C. tropicalis y números limitados de C. dubliniensis, C. inconspicua y C. guilliermondii; y Scedosporium spp., incluidos S. apiospermum, S. prolificans y Fusarium spp. Otras infecciones micóticas tratadas (a menudo con respuesta parcial o completa, véase más adelante bajo Experiencia clínica) incluyen casos aislados de Alternaria spp., Blastomyces dermatitidis, Blastoschizomyces capitatus, Cladosporium spp., Coccidioides immitis, Conidiobolus coronatus, Cryptococcus neoformans, Exserohilum rostratum, Exophiala spinifera, Fonsecaea pedrosoi, Madurella mycetomatis, Paecilomyces lilacinus, Penicillium spp. incluidos P. marneffei, Phialophora richardsiae, Scopulariopsis brevicaulis y Trichosporon spp. incluidas las infecciones por T. beigelii. Se ha observado actividad in vitro contra aislados clínicos para Acremonio spp., Alternaria spp., Bipolaris spp., Cladophialophora spp., Histoplasma capsulatum, la mayoría de las cepas de los cuales resulta inhibida por concentraciones de voriconazol en el intervalo de 0,05 a 2 mg/ml. Se ha demostrado actividad in vitro contra los siguientes patógenos, pero se desconoce la importancia clínica: Curvularia spp. y Sporothrix spp. Se deben obtener muestras para cultivo de hongos y otros estudios relevantes de laboratorio (serología, histopatología) antes de la terapia con el fin de aislar e identificar a los organismos causales. Se puede instaurar la terapia antes de conocer los resultados de los cultivos y otros estudios de laboratorio; sin embargo, una vez estos resultados estén disponibles, la terapia antiinfecciosa debe ajustarse de conformidad. Pruebas de susceptibilidad: las pruebas de susceptibilidad in vitro se llevaron a cabo conforme a los métodos del Clinical Laboratory and Standards Institute (CLSI) (M38-P para hongos filamentosos, y M27-A y M44-A para levaduras). Se han establecido los valores de corte para el voriconazol (CIM y diámetro de zona) para especies de Candida, pero no para los hongos filamentosos, incluidas las especies de Aspergillus. Métodos de las pruebas de susceptibilidad (métodos de referencia de la norma NCCLS M-27A y M44-A): nota: las pruebas de susceptibilidad por métodos de dilución exigen el uso de susceptibilidad al voriconazol en polvo. Técnicas de dilución: se usan métodos cuantitativos para determinar las concentraciones inhibitorias mínimas (CIM). Estas CIM permiten hacer estimativos de la susceptibilidad de las especies de Candida a los agentes antimicóticos. Las CIM se deben determinar usando un procedimiento estandarizado. Los procedimientos estandarizados se basan en un método de dilución (caldo) o equivalente con concentraciones estandarizadas del inóculo y concentraciones estandarizadas de voriconazol en polvo. Los valores de CIM deben ser interpretados conforme a los criterios que se dan en la siguiente tabla. Técnicas de difusión: los métodos cualitativos que exigen la medición de los diámetros de zona también ofrecen estimativos reproducibles de la susceptibilidad de las especies de Candida a un agente antimicótico. Uno de esos procedimientos estandarizados exige el uso de concentraciones estandarizadas del inóculo. En este procedimiento se usan discos de papel impregnados con 1 microgramo de voriconazol para examinar la susceptibilidad de las levaduras al voriconazol. Los criterios de interpretación de la difusión en disco también se dan en la siguiente tabla.
La categoría susceptible implica que los aislados resultan inhibidos por las concentraciones usualmente alcanzables del agente antimicótico examinado cuando se usa la dosificación recomendada para el sitio de la infección. La categoría susceptible dependiendo de la dosis implica que una infección debida al aislado puede ser tratada adecuadamente en lugares del cuerpo en los cuales los fármacos alcanzan concentraciones fisiológicas o cuando se usa una dosis más alta del fármaco. La categoría resistente implica que los aislados resultan inhibidos por las concentraciones del agente usualmente alcanzables con esquemas normales de dosificación y la eficacia clínica del agente contra el aislado no ha sido demostrada de forma fiable en estudios de tratamiento. Nota 2: estos estándares de interpretación para el voriconazol contra especies de Candida sólo son aplicables a las pruebas que se hacen con el método M27 de referencia CLSI de microdilución en caldo para CIM o el método de referencia M44 para difusión en disco para el diámetro de zona. Control de calidad (métodos estándar de referencia CLSI M-27A y M44-A): los procedimientos estandarizados de las pruebas de susceptibilidad exigen el uso de organismos de control de calidad para controlar los aspectos técnicos de los procedimientos de prueba. El voriconazol estándar en polvo y los discos de 1 mg deben arrojar el intervalo de valores señalado en la tabla que se presenta más adelante. Nota: los microorganismos de control de calidad son cepas específicas de organismos que tienen propiedades biológicas intrínsecas relacionadas con mecanismos de resistencia y su expresión genética dentro del hongo; las cepas específicas empeladas para el control microbiológico no son clínicamente significativas.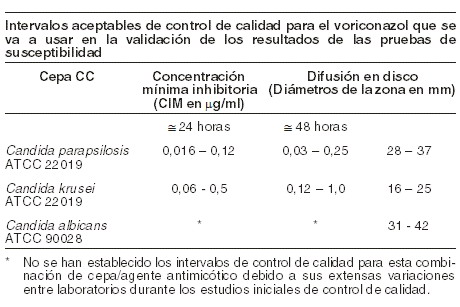
Experiencia clínica: el desenlace exitoso se define en esta sección como respuesta completa o parcial. Infecciones por Aspergillus - Eficacia en pacientes con aspergilosis de mal pronóstico: el voriconazol tiene actividad fungicida in vitro contra Aspergillus spp. La eficacia e impacto en la sobrevida, del voriconazol comparado con la amfotericina B convencional, en el tratamiento primario de aspergilosis invasiva aguda, se demostraron en un estudio abierto, aleatorizado, multicéntrico, con 277 pacientes inmunocomprometidos, tratados durante 12 semanas. Se observó una respuesta global satisfactoria (resolución completa o parcial de todos los síntomas, signos, anormalidades radiográficas/broncoscópicas atribuibles presentes en condiciones iniciales) en 53% de los pacientes tratados con voriconazol comparado con 31% de los pacientes tratados con el comparador. La tasa de supervivencia a 84 días en el grupo tratado con voriconazol fue significativamente más alta estadísticamente que la observada con el comparador y se demostró un beneficio clínicamente y estadísticamente significativo en favor del voriconazol tanto para el tiempo transcurrido hasta la muerte, como para el tiempo hasta la descontinuación del tratamiento por toxicidad. Este estudio confirmó los hallazgos de un estudio anterior de diseño prospectivo en el cual hubo desenlace positivo en sujetos con factores de riesgo de mal pronóstico, como enfermedad de injerto contra huésped, y en especial infecciones cerebrales (normalmente asociadas con una mortalidad cercana al 100%). Los estudios incluyeron aspergilosis cerebral, sinusal, pulmonar y diseminada en pacientes con trasplantes de médula ósea y órganos sólidos, enfermedades malignas hematológicas, cáncer y sida. Infecciones invasivas graves por Candida. Eficacia en pacientes no neutropénicos: la eficacia del voriconazol comparado con el régimen de amfotericina B seguido de fluconazol en el tratamiento primario de la candidemia quedó demostrada en un estudio abierto, comparativo. En este estudio se incluyeron 370 pacientes no neutropénicos con candidemia demostrada (hemocultivo positivo y signos clínicos de infección), de los cuales 248 fueron tratados con voriconazol. La población de pacientes estaba gravemente enferma, aproximadamente 50% de los sujetos en la unidad de cuidado intensivo y 40% en respirador en condiciones iniciales. La media de la duración del tratamiento fue de 15 días en los dos grupos de tratamiento. Se observó una respuesta exitosa (resolución/mejoría de todos los signos y síntomas clínicos de infección, hemocultivos negativos para Candida, sitios de infección tisular profunda negativos para Candida) en 41% de los pacientes en los dos grupos de tratamiento 12 semanas después del final de la terapia (FDT). En este análisis, los pacientes a los que no se les hizo valoración 12 semanas después del final de la terapia (FDT) fueron clasificados como fracasos. De acuerdo a un análisis secundario, en el cual se compararon las tasas de respuesta en el intervalo de tiempo más tardío y más relevante para la evaluación del paciente (FDT, o 2, 6, o 12 semanas después del FDT), el voriconazol y la terapia de amfotericina B seguida de fluconazol tuvieron tasas de respuesta de 65% y 71%, respectivamente. Infecciones refractarias graves por Candida: el estudio comprendió 55 pacientes con infecciones sistémicas refractarias graves por Candida (incluidos candidemia, candidiasis diseminada y otras invasivas) en las cuales el tratamiento antimicótico previo, en especial con fluconazol, había resultado ineficaz. Se observó respuesta exitosa en 24 pacientes (15 respuestas completa, 9 parciales). En cepas de Candida no albicans resistentes al fluconazol, se observó un desenlace exitoso en 3/3 infecciones por C. krusei (respuestas completas) y 6/8 infecciones por C. glabrata (5 respuestas completas, 1 parcial). Los datos de eficacia clínica fueron respaldados por datos limitados de susceptibilidad. Otros patógenos micóticos poco frecuentes: se demostró que el voriconazol fue efectivo contra los siguientes patógenos micóticos poco frecuentes: Scedosporium spp.- se observó una respuesta exitosa a la terapia con voriconazol en 16 de 28 pacientes (55%) con infección por S. apiospermum y en 2 de 7 pacientes (29%) con infección por S. prolificans. Además, se observó una respuesta exitosa en 1 de 3 pacientes con infecciones por organismos mixtos. Fusarium spp: 7 de 17 pacientes (41%) se trataron con éxito con voriconazol. De estos 7 pacientes, 3 tenían infección ocular, 1 sinusal y 3 diseminada. Cuatro pacientes adicionales con fusariosis tenían una infección causada por varios organismos; dos de ellos tuvieron un desenlace exitoso. La mayoría de los pacientes que recibieron tratamiento con voriconazol para las infecciones raras arriba mencionadas, fue intolerante o refractaria a la terapia antimicótica previa. Duración del tratamiento: el voriconazol oral permite flexibilidad para la atención del paciente y la posibilidad de tratamiento prolongado cuando está indicado. En los estudios clínicos, 561 pacientes recibieron terapia con voriconazol durante más de 12 semanas, y 136 sujetos recibieron voriconazol durante más de 6 meses. Estudios clínicos en niños: 61 niños con edades de 9 meses hasta 15 años con infecciones micóticas invasivas definidas o probables, fueron tratados con voriconazol. Esta población incluyó 34 pacientes de 2 a < 12 años de edad y 20 pacientes 12-15 años de edad. La mayoría (57/61) tenía terapias antimicóticas previas fallidas. Los estudios terapéuticos incluyeron cinco pacientes con edades de 12-15 años, y los restantes pacientes recibieron voriconazol en programas de uso compasivo. Las enfermedades subyacentes en estos pacientes incluyeron enfermedades hematológicas malignas y anemia aplásica (27 pacientes) y enfermedad granulomatosa crónica (14 pacientes). La infección micótica más frecuentemente tratada fue aspergilosis (43/61; 70%). Estudios clínicos que examinaron el intervalo QT: se llevó a cabo un estudio controlado con placebo, aleatorizado, de dosis única, de diseño cruzado, para evaluar el efecto sobre el intervalo QT en voluntarios sanos con tres dosis orales de voriconazol y ketoconazol. La media de los aumentos máximos del QTc ajustados por placebo desde el valor inicial después de 800, 1200 y 1600 mg de voriconazol fueron de 5,1, 4,8, y 8,2 mseg, respectivamente y 7,0 mseg para el ketoconazol 800 mg. Ninguno de los sujetos de los grupos tuvo un aumento del QTc de ≥ 60 mseg desde el valor inicial. Ningún sujeto experimentó un intervalo que excediera el umbral clínica y potencialmente relevante de 500 mseg. Propiedades farmacocinéticas: características farmacocinéticas generales: la farmacocinética del voriconazol ha sido caracterizada en sujetos sanos, poblaciones especiales y pacientes. Durante la administración oral de 200 mg o 300 mg dos veces al día durante 14 días en pacientes en riesgo de aspergilosis (sobre todo pacientes con neoplasias malignas del tejido linfático o hematopoyético), las características farmacocinéticas observadas de absorción rápida y consistente, acumulación y farmacocinética no lineal concordaron con las observadas en sujetos sanos. Las variables farmacocinéticas del voriconazol es no lineal debido a la saturación de su vía metabólica. Se observa un aumento más que proporcional en la exposición al aumentar la dosis. Se calcula que, en promedio, cuando se aumenta la dosis oral de 200 mg dos veces al día a 300 mg dos veces al día se obtiene un aumento de 2,5 veces en la exposición (ABCt). Cuando se administran los regímenes recomendados de dosis intravenosa o dosis oral de carga, se alcanzan concentraciones plasmáticas cercanas a las del estado de equilibrio dentro de las primeras 24 horas de la administración. Sin la dosis de carga inicial, la acumulación ocurre durante la dosificación múltiple dos veces al día alcanzando concentraciones plasmáticas de voriconazol en estado estable en el día 6, en la mayoría de los sujetos. Distribución: el volumen de distribución en el estado estable del voriconazol se estima en 4,6 l/kg, lo cual sugiere una extensa distribución dentro de los tejidos. La unión a las proteínas plasmáticas se calcula en 58%. Todas las muestras de líquido cefalorraquídeo de ocho pacientes en un programa compasivo mostraron concentraciones detectables de voriconazol. Metabolismo: en estudios in vitro se ha demostrado que el voriconazol es metabolizado por las isoenzimas hepáticas del citocromo P450, CYP2C19, CYP2C9 y CYP3A4. La variabilidad de la farmacocinética del voriconazol entre individuos es alta. Los estudios in vivo indicaron que la CYP2C19 desempeña un papel clave en el metabolismo del voriconazol. Esta enzima exhibe polimorfismo genético. Por ejemplo, es de esperar que 15-20% de las poblaciones asiáticas sean metabolizadoras lentos. Entre blancos y negros, la prevalencia de metabolizadores lentos es de 3-5%. Los estudios realizados en sujetos sanos de raza blanca y japoneses demostraron que los metabolizadores lentos tienen en promedio una exposición 4 veces más alta al voriconazol (ABCt) que sus contrapartes homocigotos metabolizadoras rápidos. Los sujetos heterocigotos con metabolismo extenso rápidos tienen en promedio una exposición 2 veces más alta al voriconazol que sus contrapartes metabolizadores rápidos homocigotos. El principal metabolito del voriconazol es el N-óxido, que que corresponde al 72% de los metabolitos radiomarcados circulantes en el plasma. Este metabolito tiene mínima actividad antimicótica y no contribuye a la eficacia general del voriconazol. Excreción: el voriconazol se elimina a través del metabolismo hepático y menos de 2% de la dosis se excreta sin cambios por la orina. Después de la administración de una dosis radiomarcada de voriconazol, aproximadamente 80% de la radioactividad se recupera en la orina después de múltiples dosis intravenosas y 83% en la orina después de múltiples dosis orales. La mayoría ( > 94%) de la radioactividad total se excreta en las primeras 96 horas después de las dosis orales o intravenosas. La vida media terminal del voriconazol depende de la dosis y es de aproximadamente 6 horas luego de 200 mg (por vía oral). A causa de la farmacocinética no lineal, la vida media terminal no es útil para la predicción de la acumulación o eliminación del voriconazol. Relaciones farmacocinéticas-farmacodinámicas (FC/FD): en 10 estudios terapéuticos, las concentraciones plasmáticas promedio y máxima en los individuos de los estudios fueron de 2425 ng/ml (intervalo entre cuartiles 1193 a 4380 ng/ml) y 3742 ng/ml (intervalo entre cuartiles 2027 a 6302 ng/ml), respectivamente. No se encontró una asociación positiva entre la concentración plasmática media, máxima o mínima de voriconazol y la eficacia en estudios terapéuticos. Los análisis FC/FD de los datos de los estudios clínicos identificaron asociaciones positivas entre las concentraciones plasmáticas del voriconazol y las anormalidades de las pruebas de función hepática y los trastornos visuales. Farmacocinética en grupos especiales de pacientes: género: en un estudio de múltiples dosis orales, la Cmáx y el ABCt en mujeres jóvenes sanas fueron 83% y 113% más altas, respectivamente, que en hombres jóvenes sanos (18-45 años), después de la dosificación en tableta. En el mismo estudio no se observaron diferencias significativas en la Cmáx y el ABCt entre hombres ancianos sanos y mujeres ancianas sanas (≥65 años). En un estudio similar, después de dar la suspensión oral, la media del ABCt en mujeres jóvenes sanas fue 45% más alta que en hombres jóvenes sanos, en tanto que la media de la Cmáx fue comparable entre los sexos. Las concentraciones de voriconazol en el estado estable (Cmín) observadas en mujeres fueron 100% y 91% más altas que las observadas en varones que recibieron la tableta y la suspensión oral, respectivamente. En el programa clínico no se hizo ajuste de la dosificación con base en el sexo. El perfil de seguridad y las concentraciones plasmáticas observadas en pacientes de ambos sexos fueron similares. Por ello, no es necesario hacer ajustes de la dosificación con base en el género. Ancianos: en un estudio con múltiples dosis orales la Cmáx y el ABCt de hombres ancianos sanos (≥65 años) fueron 61% y 86% más altos, respectivamente, que en hombres jóvenes sanos (18-45 años). No se observaron diferencias significativas en la Cmáx y el ABCt entre mujeres ancianas sanas (≥65 años) y mujeres jóvenes sanas (18-45 años). En los estudios terapéuticos no se hizo ajuste de la dosificación con base en la edad. Se observó una relación entre las concentraciones plasmáticas y la edad. Sin embargo, el perfil de seguridad del voriconazol en pacientes jóvenes y viejos fue similar y, por ello, no es necesario hacer ajustes de la dosificación en los ancianos. Población pediátrica: la dosis intravenosa recomendada en los pacientes pediátricos está basada en los datos combinados de un análisis farmacocinético poblacional en 82 pacientes pediátricos inmunocomprometidos, con edades de 2 a < 12 años, quienes se evaluaron en tres estudios farmacocinéticos (examinando dosis intravenosas únicas de 3 y 4mg/kg dos veces al día, dosis intravenosas múltiples de 3, 4, 6 y 8 mg/kg dos veces al día y dosis múltiples de suspensión oral de 4 y 6 mg/kg dos veces al día). La mayoría de los pacientes recibieron más de un nivel diferente de dosis, con una duración máxima de dosificación de 30 días. Una comparación de los datos farmacocinéticos de las poblaciones pediátrica y adulta, indicó que a fin de obtener exposiciones comparables a las obtenidas en los adultos después de dosis de mantenimiento de 4 mg/kg dos veces al día, en los pacientes pediátricos se requerían dosis intravenosas de mantenimiento de 7 mg/kg dos veces al día. La dosis intravenosa de mantenimiento más alta en los pacientes pediátricos, en comparación con la de los adultos, refleja la mayor capacidad de eliminación en los pacientes pediátricos, debido a su mayor proporción de masa hepática/masa corporal. Para obtener exposiciones comparables a las de los adultos después de dosis intravenosas de mantenimiento de 3 mg/kg dos veces al día, en los pacientes pediátricos se requieren dosis intravenosas de mantenimiento de 4 mg/kg dos veces al día. Con base en los resultados del análisis farmacocinético poblacional, no se justifica dosis de carga o un ajuste de la dosificación por la edad, en los pacientes con edades de 2 a < 12 años. Deterioro de la función renal: en un estudio con dosis única oral (200 mg) en sujetos con función renal normal y deterioro leve (depuración de creatinina 41-60 ml/min) a grave (depuración de creatinina < 20 ml/min) de la función renal, la farmacocinética del voriconazol no se vio afectada en grado significativo por el deterioro de la función renal. La unión del voriconazol a las proteínas plasmáticas fue similar en sujetos con diferentes grados de deterioro de la función renal. (Ver Precauciones generales y Dosis y vía de administración). En pacientes con disfunción renal moderada a grave (niveles de creatinina sérica ≥ 220 micromol/l - 2,5 mg/dl), se presenta la acumulación del vehículo intravenoso, SBECD. (ver Precauciones generales y Dosis y vía de administración). Deterioro de la función hepática: después de una dosis única oral (200 mg), el ABCt fue 233% más alto en sujetos con cirrosis hepática leve a moderada (Child-Pugh A y B) comparados contra sujetos con función hepática normal. La unión del voriconazol a las proteínas no resultó afectada por el deterioro de la función hepática. En un estudio de múltiples dosis orales, el ABCt fue similar en sujetos con cirrosis hepática moderada (Child-Pugh B) que recibieron dosis de mantenimientos de 100 mg dos veces al día y sujetos con función hepática normal que recibieron 200 mg dos veces al día. No hay datos farmacocinéticos disponibles de pacientes con cirrosis hepática grave (Child-Pugh C). Si desea conocer la información sobredosificación (ver Dosis y vía de administración).
Contraindicaciones: el voriconazol está contraindicado en pacientes con hipersensibilidad conocida al voriconazol o a cualquiera de los excipientes. La administración concomitante de los sustratos CYP3A4, terfenadina, astemizol, ciprasida, pimozida o quinidina con voriconazol, está contraindicada debido a que las concentraciones plasmáticas incrementadas de estos productos medicinales, pueden llevar a la prolongación del QTc y casos poco probables de Torsade de Pointes (ver Interacciones medicamentosas y de otro género). La administración concomitante de voriconazol y sirolimus está contraindicada, debido a que se ha demostrado que el voriconazol aumenta significativamente las concentraciones plasmáticas de sirolimus en sujetos sujetos sanos (ver Interacciones medicamentosas y de otro género). La administración concomitante de voriconazol con rifampicina, carbamazepina o barbitúricos de acción prolongada (p. ej. fenobarbital), está contraindicada debido a que es probable que estos productos medicinales disminuyan de manera significativa las concentraciones plasmáticas de voriconazol (ver Interacciones medicamentosas y de otro género). La administración concomitante de voriconazol con altas dosis de ritonavir (más de 400 mg dos veces al día) está contraindicada debido a que el ritonavir, en esta dosis, disminuyó significativamente las concentraciones plasmáticas de voriconazol en sujetos sanos (ver Interacciones medicamentosas y de otro género, para dosis más bajas, ver Precauciones generales). La administración concomitante de alcaloides del cornezuelo de centeno (ergotamina, dihidroergotamina) los cuales son sustratos del CYP3A4, está contraindicada debido a que las concentraciones plasmáticas aumentadas de estos productos medicinales pueden conducir a ergotismo (ver Interacciones medicamentosas y de otro género). La administración concomitante de voriconazol con la hierba de San Juan está contraindicada (ver Interacciones medicamentosas y de otro género).
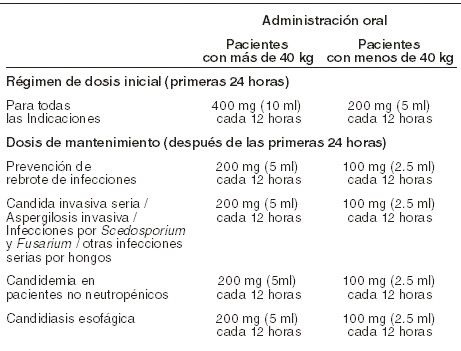
Precauciones generales: Hipersensibilidad: es preciso tener precaución al prescribir voriconazol a pacientes con hipersensibilidad a otros azoles. Eventos adversos cardíacos: algunos azoles, incluyendo el voriconazol, han sido asociados con la prolongación del intervalo QT. Durante el desarrollo clínico y la vigilancia post-comercialización se han presentado algunos casos de torsades de pointes en pacientes que toman voriconazol. Estos casos involucraron a pacientes gravemente enfermos con múltiples factores de riesgo que pueden prestarse a confusión, como antecedentes de quimioterapia cardiotóxica, cardiomiopatía, hipokalemia y medicaciones concomitantes que pueden haber contribuido. Voriconazol deberá administrarse con precaución a pacientes con estas afecciones potencialmente proarrítmicas (ver Dosis y vía de administración). Se ha realizado un estudio en voluntarios sanos examinando el efecto sobre el intervalo QT de dosis únicas de voriconazol de hasta 4 veces la dosis diaria usual. Ninguno de los sujetos en ninguno de los grupos tuvo prolongación del QTc ≥ 60 mseg desde el valor basal. Ningún sujeto experimentó un intervalo que superase el umbral clínica y potencialmente relevante de 500 mseg (ver Farmacocinética y farmacodinamia, Propiedades farmacodinámicas). Toxicidad hepática: en estudios clínicos se han presentado pocos casos (≥0,1% y < 1%) de reacciones hepáticas graves durante el tratamiento con voriconazol (incluidos hepatitis clínica, colestasis e insuficiencia hepática fulminante, aun con casos fatales). Se observaron casos de reacciones hepáticas, tienen lugar sobre todo en pacientes que tienen padecimientos médicos graves subyacentes (principalmente enfermedades malignas de origen hematológico). Las reacciones hepáticas transitorias, incluidas hepatitis e ictericia, han sobrevenido en pacientes que no tienen otros factores de riesgo identificables. La disfunción hepática por lo general ha sido reversible al suspender la terapia. Monitoreo de la función hepática: se recomienda la vigilancia habitual de la función hepática de los pacientes que se encuentran en terapia con voriconazol, en especial mediante pruebas de función hepática y bilirrubinas. Se deberá considerar la suspensión del voriconazol si aparecen signos y síntomas clínicos concordantes con enfermedad hepática que puedan ser atribuibles al voriconazol (ver sección Dosis y vía de administración). Eventos adversos visuales: han habido informes post-comercialización de eventos adversos visuales prolongados, incluyendo neuritis óptica y papiledema. Estos eventos se produjeron principalmente en pacientes gravemente enfermos que habían estado en condiciones subyacentes y/o tomando concomitantemente medicamentos que pudieran haber causado o contribuido a éstos (ver Dosis y vía de administración). Eventos adversos renales: se ha observado insuficiencia renal aguda en pacientes gravemente enfermos sometidos a tratamiento con voriconazol. Es probable que los pacientes que están siendo tratados con voriconazol también reciban al mismo tiempo medicaciones nefrotóxicas o que tengan padecimientos concurrentes que pueden dar lugar a deterioro de la función renal. Monitoreo de la función renal: se debe vigilar el desarrollo de anormalidades de la función renal. Esto debe incluir evaluación de laboratorio, en especial de la creatinina sérica (ver Dosis y vía de administración). Monitoreo de la función pancreática: será preciso monitorear a los adultos y niños con factores de riesgo de pancreatitis aguda (p.ej., quimioterapia reciente, transplante de células madre hematopoyéticas [HSCT]), hay que vigilar el desarrollo de la pancreatitis durante el tratamiento con voriconazol. Eventos adversos dermatológicos: en raras ocasiones algunos pacientes han desarrollado reacciones cutáneas exfoliativas, entre ellas síndrome de Stevens-Johnson, durante el tratamiento con voriconazol. En el caso de que un paciente desarrolle una reacción cutánea exfoliativa será preciso suspender la administración de voriconazol. Además, voriconazol se ha asociado con reacción cutánea de fotosensibilidad. Se recomienda que los pacientes eviten la exposición intensa o prolongada a la luz solar directa durante el tratamiento con voriconazol. Se ha reportado carcinoma de células epidermoides de la piel y melanoma durante la terapia prolongada. En el caso de que un paciente desarrolle una lesión cutánea consistente con carcinoma de células epidermoides o melanoma, será preciso considerar la suspensión de voriconazol. Uso en niños: no se han establecido la seguridad y efectividad en niños menores de dos años de edad (ver Farmacocinética y farmacodinamia - Propiedades farmacodinámicas). El voriconazol está indicado en niños mayores de dos años de edad. Se debe vigilar la función hepática tanto en niños como en adultos. La biodisponibilidad oral puede ser limitada en niños de 2 a 12 años con mala absorción y peso corporal muy bajo para la edad. En ese caso, se recomienda la administración intravenosa del voriconazol. Metadona (sustrato de CYP3A4): las concentraciones plasmáticas elevadas de la metadona se han asociado con toxicidad incluida la prolongación del QT. Se recomienda la vigilancia frecuente de eventos adversos y toxicidad relacionada con la metadona durante la co-administración. Se puede necesitar reducir la dosis de metadona (ver Interacciones medicamentosas y de otro género). Opiáceos de acción breve (sustrato de CYP3A4): se deberá considerar la reducción de la dosis de alfentanil y otros opiáceos de acción breve con estructura similar a la de alfentanil y metabolizados por CYP3A4 (e.g., sufentanil, fentanil) cuando se administren concomitantemente con voriconazol (ver sección Interacciones medicamentosas y de otro género). Tomando en consideración que la vida media de alfentanil se prolonga cuando el mencionado agente se administra concomitantemente con voriconazol, pudiera ser necesario realizar un monitoreo frecuente por eventos adversos asociados con los opiáceos (incluyendo un período más prolongado de monitoreo respiratorio. Oxicodona (sustrato de CYP3A4): será preciso considerar la reducción de la dosis de oxicodona y otros opiáceos de acción prolongada metabolizados por CYP3A4 (e.g., hidrocodona) cuando se administren concomitantemente con voriconazol. Puede ser que se necesite realizar un monitoreo frecuente por eventos adversos asociados con los opiáceos (ver sección Interacciones medicamentosas y de otro género). Fenitoína (sustrato de CYP2C9 y potente inductor de CYP450): se recomienda ejercer una estrecha vigilancia de niveles de fenitoína cuando se administran fenitoína con voriconazol al mismo tiempo. Se deberá evitar el uso concomitante de voriconazol y fenitoína a menos que el beneficio supere al riesgo (ver Interacciones medicamentosas y de otro género). Rifabutina (inductor de CYP450): se recomienda ejercer una estrecha vigilancia de cuadro hemático completo y eventos adversos del rifabutina (p.ej., uveítis) cuando se administra rifabutina con voriconazol, al mismo tiempo. Deberá evitarse el uso concomitante de voriconazol y rifabutina a menos que el beneficio supere al riesgo (ver Interacciones medicamentosas y de otro género). Ritonavir: deberá evitarse la co-administración de voriconazol y ritonavir en bajas dosis (100 mg dos veces al día) a menos que el beneficio/riesgo justifique el uso del voriconazol. (ver sección Interacciones medicamentosas y de otro género, para las dosis más altas, ver Contraindicaciones). Efavirenz (inductor de CYP450; inhibidor y sustrato de CYP3A4): cuando el voriconazol se administra al mismo tiempo con efavirenz se debe aumentar la dosis de voriconazol a 400 mg dos veces al día y disminuir la de efavirenz a 300 mg una vez al día (ver Interacciones medicamentosas y de otro género). Efectos sobre la capacidad de conducir vehículos y operar maquinarias: el voriconazol puede causar cambios transitorios y reversibles de la visión, incluidos visión borrosa, alteración/reforzamiento de la percepción visual, o fotofobia. Los pacientes tienen que evitar las tareas potencialmente riesgosas, como conducir vehículos u operar maquinarias mientras experimentan estos síntomas. Los pacientes no deben conducir por la noche mientras toman voriconazol.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no hay información adecuada disponible sobre el uso de voriconazol en mujeres embarazadas. En estudios con animales se ha demostrado la toxicidad reproductiva en dosis altas (ver Precauciones y Relación con efecto de carcinogénesis, mutagenésis, teratogenésis y sobre fertilidad). El riesgo potencial en seres humanos es desconocido. No se deberá usar voriconazol durante el embarazo a menos que el beneficio para la madre claramente supere el riesgo portencial para el feto. Las mujeres con posibilidad de concebir deberán siempre usar anticoncepción efectiva durante el tratamiento. Lactancia: no se ha investigado la excreción del voriconazol en la leche materna. No se deberá usar el voriconazol en madres lactantes a menos que el beneficio claramente supere el riesgo.
Reacciones secundarias y adversas: El perfil de seguridad del voriconazol se basa en una base de datos de seguridad integrada de más de 2000 sujetos (1655 pacientes en estudios terapéuticos). Esto representa una población heterogénea, que contiene pacientes con enfermedad maligna hematológica, pacientes con infección por VIH con candidiasis esofágica e infecciones micóticas refractarias, pacientes no neutropénicos con candidemia o aspergilosis y voluntarios sanos. 561 pacientes tuvieron una terapia con voriconazol de más de 12 semanas de duración, y 136 pacientes recibieron voriconazol durante más de 6 meses. La siguiente tabla incluye reacciones adversas de estudios terapéuticos y/o compasivos/extensivos, si es que existen posibles relaciones de causalidad. Los eventos adversos informados con mayor frecuencia fueron trastornos visuales, fiebre, erupción cutánea, vómito, náuseas, diarrea, dolor de cabeza, edema periférico y dolor abdominal. La gravedad de los eventos adversos en general fue leve a moderada. No se observaron diferencias clínicamente significativas al analizar los datos de seguridad por edad, raza, género sexual.
Trastornos visuales: los trastornos visuales relacionados con el tratamiento con voriconazol fueron muy frecuentes. En ensayos terapéuticos, aproximadamente 21% de los sujetos experimentó alteración/reforzamiento de la percepción visual, visión borrosa, cambios de la visión del color o fotofobia. Los trastornos visuales son transitorios y completamente reversibles, ya que la mayoría se resuelve espontáneamente en 60 minutos. Hay evidencia de atenuación con las dosis repetidas de voriconazol. El trastorno visual suele ser leve, rara vez obliga a la interrupción del medicamento y no se ha asociado con secuelas de largo plazo. Los trastornos visuales se pueden asociar con niveles plasmáticos dosis-elevados. Se han recibido reportes posteriores al mercadeo de eventos adversos visuales prolongados (ver Precauciones Generales). Se desconoce el mecanismo de acción, aunque lo más probable es que el sitio de la acción se encuentre dentro de la retina. En un estudio de voluntarios sanos en el cual se investigó el impacto de voriconazol sobre la función retiniana, voriconazol ocasionó un descenso en la amplitud de la forma de las ondas en el electroretinograma (ERG). El ERG mide las corrientes eléctricas en la retina. Los cambios en el ERG no progresaron a lo largo de 29 días de administración y fueron completamente reversibles al suspenderse la administración de voriconazol. Se evaluó el efecto a largo plazo de voriconazol (mediana 169 días; rango 5-353 días) sobre la función visual en sujetos con paracoccidioidomicosis. Voriconazol no exhibió ningún efecto clínicamente relevante sobre la función visual según se evaluó a través de pruebas de agudeza visual, campos visuales, visión del color y sensibilidad ante el contraste. No se percibieron signos de toxicidad retiniana. 17/35 sujetos medicados con voriconazol experimentaron eventos adversos visuales. Dichos eventos no ocasionaron la suspensión de la terapia, por lo general fueron leves, se presentaron durante la primera semana de la terapia y se resolvieron durante la terapia continua con voriconazol. Reacciones dermatológicas: las reacciones dermatológicas fueron frecuentes en los pacientes tratados con voriconazol en los estudios clínicos, pero estos pacientes tenían enfermedades subyacentes serias y estaban recibiendo múltiples medicaciones concomitantes. La mayoría de las erupciones cutáneas fueron leves a moderadas. Los pacientes han sufrido reacciones cutáneas serias, incluidos síndrome de Stevens-Johnson (infrecuente), necrolisis epidérmica tóxica (rara) y eritema multiforme (rara) durante el tratamiento con voriconazol. Si un pacientes presenta una erupción cutánea debe ser sometido a estrecha vigilancia y suspender el voriconazol si las lesiones avanzan. Los pacientes que reciben terapia a largo plazo con voriconazol han presentado reacciones de fotosensibilidad (ver Precauciones generales). Pruebas de función hepática: la incidencia global de anormalidades clíni